女,6岁4月龄,因"夜间睡眠不能平卧,伴张口呼吸4个月余,加重2个月"于2015年12月入住首都医科大学附属北京儿童医院。入院前4个月,患儿出现夜间睡眠不能平卧,伴张口呼吸,无打鼾、气促、喘憋、咳嗽、咳痰、头晕等表现,日间自觉有乏力,运动后明显,体力、食量少于同龄儿童,喜静。入院前2个月,上述症状加重,行超声心动图提示肺动脉高压、右心室肥厚。外院多导睡眠监测(PSG)诊断重度阻塞性睡眠呼吸暂停综合征(OSAS),予持续正压通气(CPAP)治疗无好转。患儿为第1胎,足月自然生产,5月龄竖头,8月龄独坐,15月龄独走,自幼跑跳差,能上下楼梯、较慢,智力正常。无高血压、糖尿病、神经肌肉病家族史。1岁患肺炎,5岁患重症肺炎。
体格检查:神志清,反应好,发育正常,无特殊面容,身高117 cm,体重20.0 kg,呼吸30次/min,脉搏108次/min,双侧扁桃体Ⅱ度。心、肺、腹查体未见明显异常。步态无异常,Gower征(+),轻度翼状肩胛,脊柱前凸并脊柱强直,四肢肌张力偏低,肌力轻度下降,近端明显,为Ⅳ+,远端Ⅴ-至Ⅴ,双侧膝腱反射减弱。辅助检查:血常规:白细胞9.0×109/L,红细胞4.64×1012/L,血红蛋白125 g/L,血小板320×109/L,中性粒细胞0.51,淋巴细胞0.36。胸X线片:双肺纹理稍粗,心膈未见异常。超声心动图:轻-中度肺动脉高压,右心室肥厚。肺功能:因患儿不能配合未完成。头颅磁共振成像(MRI)未见异常;血清肌酸激酶正常;肌电图未见肌源性或神经源性损害的特征性改变;下肢肌肉MRI:双侧臀大肌重度脂肪浸润伴萎缩。当夜PSG及同步经皮CO2监测显示:患儿睡眠中无打鼾,无矛盾呼吸,呼吸事件以通气不足为主要表现,平均经皮血氧饱和度(SpO2):0.90,最低0.73;平均经皮二氧化碳(EtCO2):74 mmHg(1 mmHg=0.133 kPa),最高86 mmHg,EtCO2>50 mmHg的时间占总睡眠时间的96.2%。PSG趋势图见图1。
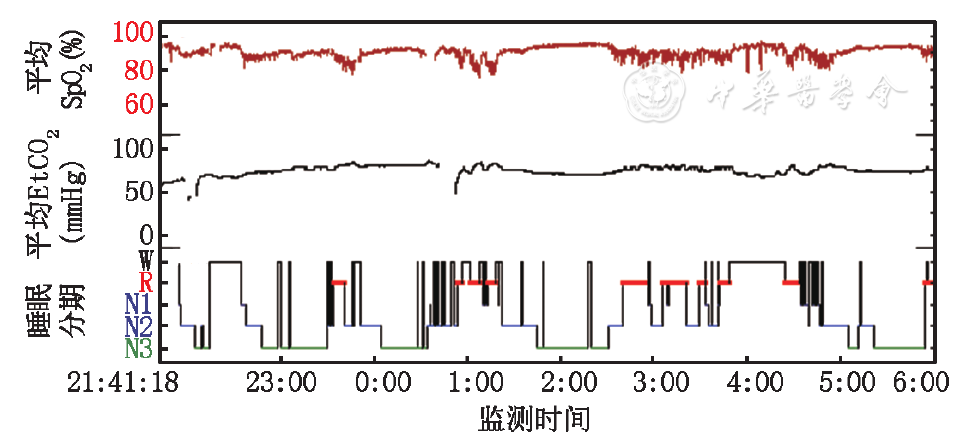
SpO2:经皮血氧饱和度;EtCO2:经皮二氧化碳;W:清醒期;R:快眼动睡眠期;N1:非快眼动睡眠期1期;N2:非快眼动睡眠期2期;N3:非快眼动睡眠期3期;1 mmHg=0.133 kPa
基因检查:北京康旭医学检验所进行患儿及其父母基因测序,SEPN1基因发现c.2_3insGGGCC、c.3_4insGGCCGGGCCC的杂合核苷酸变异。患儿上述变异分别遗传自父母,其父母均只携带其中一个杂合变异(图2)。
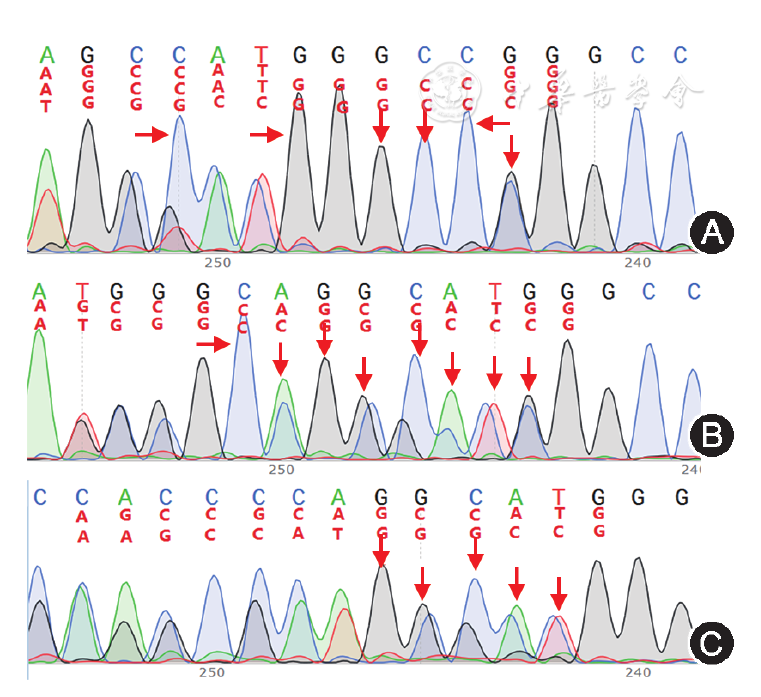
箭头为基因变异位点
采用手工压力滴定并对睡眠、呼吸及经皮CO2行同步监测。使用鼻罩,采用双水平自主/时间控制可切换模式(S/T模式),吸气压力与呼气压力分别为14和4 cmH2O (1 cmH2O=0.098 kPa),治疗开始后,患儿平均SpO2 0.94,平均EtCO2 72 mmHg (图3) 。3个月后进行呼吸机压力再次调试,吸气压力与呼气压力分别为14和5 cmH2O,平均SpO2 0.98,平均EtCO2 66 mmHg (图4)。
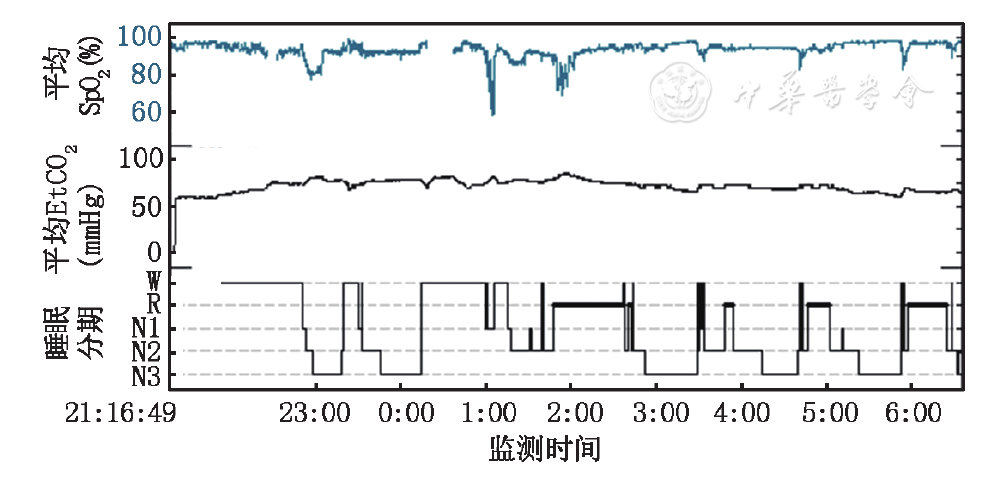
SpO2:经皮血氧饱和度;EtCO2:经皮二氧化碳;W:清醒期;R:快眼动睡眠期;N1:非快眼动睡眠期1期;N2:非快眼动睡眠期2期;N3:非快眼动睡眠期3期;1 mmHg=0.133 kPa
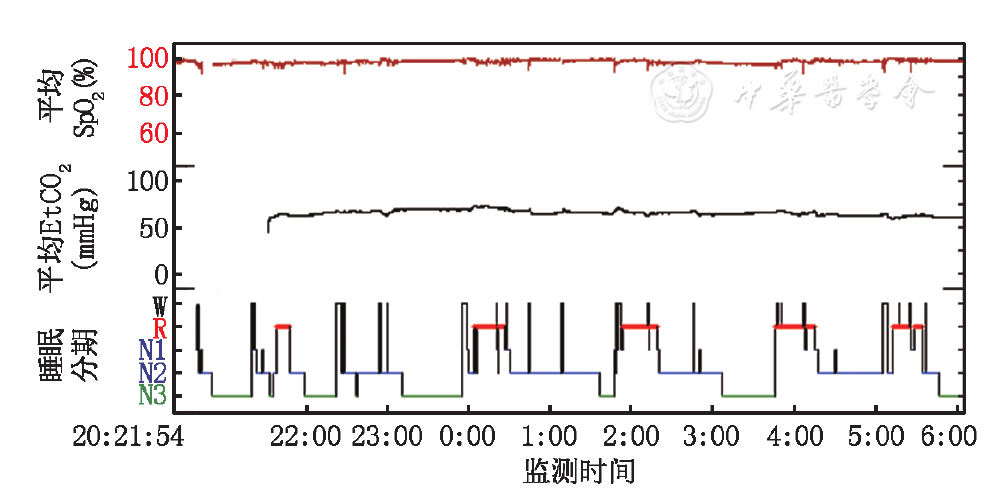
SpO2:经皮血氧饱和度;EtCO2:经皮二氧化碳;W:清醒期;R:快眼动睡眠期;N1:非快眼动睡眠期1期;N2:非快眼动睡眠期2期;N3:非快眼动睡眠期3期;1 mmHg=0.133 kPa
呼吸机治疗半年后电话随访,患儿夜间睡眠好,人机同步好,晨起无头痛、乏力。复查超声心动,肺动脉高压及右心室肥大较前好转。
本例患儿入睡后出现肺泡通气不足,经整夜PSG及同步EtCO2监测显示患儿有睡眠中呼吸运动变浅、通气不足、持续低氧血症和CO2潴留,EtCO2>50 mmHg的时间占总睡眠时间的百分比超过25%,根据国际睡眠疾病分类第三版(ICSD-3)[1]诊断为睡眠相关低通气疾病。结合患儿病史、查体及基因检查SEPN1基因变异,故确诊为SEPN1基因相关肌病。如果不予治疗,可能出现红细胞增多症、肺动脉高压、心功能不全等,因此,应早期诊断和治疗,以避免严重并发症的发生。由于神经肌肉病患者早期的呼吸障碍不是呼吸衰竭,而是夜间低通气,因此PSG已被推荐作为评估神经肌肉病患者呼吸障碍的重要方法[2]。在不明原因的肺动脉高压及右心室肥大的患者,也应注意低通气的可能。本例患儿存在肺动脉高压、右心室肥厚,PSG显示夜间入睡后SpO2持续下降,没有打鼾等阻塞性呼吸事件的表现,加之EtCO2监测结果明确了诊断。患儿在门诊诊治,未能行血气检测,由于夜间连续血气检测的不可行性,国际睡眠研究会指出,EtCO2监测可作为替代方法诊断低通气疾病;此外,因患儿呼吸肌肉无力,不能完成肺功能吸气再用力呼气的动作,未能完成肺功能检查,是本病例遗憾之处。
SEPN1基因相关肌病病程缓慢,主要表现为生长发育过程中肌张力低下、大运动发育落后、脊柱强直、活动耐力下降、呼吸功能障碍等。由于患者早期运动发育基本正常,四肢活动没有明显无力表现,进行性肌肉病变的证据有限(肌酸激酶正常或轻度升高),且肌病过程相对缓慢[3],往往容易被家长和临床医生忽视。但是,患者入睡后呼吸肌群无力、呼吸中枢敏感性下降明显,因此,睡眠相关低氧血症主要发生于睡眠期,这种夜间的血气异常可以导致患者心血管和神经认知损害,甚至猝死,值得重视。基因检查可明确SEPN1基因相关肌病的诊断。本例患儿自幼有活动耐力下降、易疲劳表现,考虑神经肌肉病,SEPN1基因检测发现c.2_3insGGGCC、c.3_4insGGCCGGGCCC的复合杂合变异。在蛋白质水平,上述变异分别导致从第5号氨基酸Arg开始的氨基酸合成发生改变,并在改变后的第63个氨基酸终止(p.Arg5GlyfsTer63)、从第8号氨基酸Gln开始的氨基酸合成发生改变,并在改变后的第78个氨基酸终止(p.Gln8ProfsTer78),均为移码变异,均可能导致蛋白质功能受到影响,均不属于多态性变化(参考数据库:1000Genomes、dbSNP)。受检者上述变异分别遗传自父母(均只携带其中一个杂合变异),符合常染色体隐性遗传病遗传方式。上述变异均尚未见文献报道(参考数据库:HGMD Pro及PubMed)。
睡眠低通气患者的呼吸异常表现为中枢性低通气,因此,需要采用双水平呼吸机的模式[4]。Annane等[4]回顾了41例SEPN1基因相关肌病的临床资料,在15岁时,50%的患儿需要无创呼吸支持,而到20岁时这一比例增加到了75%。本患儿6岁时确诊,已经出现严重的夜间低氧血症和CO2潴留,并合并肺动脉高压和右心室肥厚,表明不同疾病个体呼吸功能损害的进程不同,定期的监测、及时的干预非常重要。无创通气治疗在神经肌肉病引起的睡眠相关低通气的患儿能够成功实施,并达到满意的治疗效果,从而减缓神经肌肉病患儿疾病的进程,避免其早期即出现呼吸、循环衰竭,减少了医疗负担[5]。

女,6岁4月龄,因"夜间睡眠不能平卧,伴张口呼吸4个月余,加重2个月"于2015年12月入住首都医科大学附属北京儿童医院。入院前4个月,患儿出现夜间睡眠不能平卧,伴张口呼吸,无打鼾、气促、喘憋、咳嗽、咳痰、头晕等表现,日间自觉有乏力,运动后明显,体力、食量少于同龄儿童,喜静。入院前2个月,上述症状加重,行超声心动图提示肺动脉高压、右心室肥厚。外院多导睡眠监测(PSG)诊断重度阻塞性睡眠呼吸暂停综合征(OSAS),予持续正压通气(CPAP)治疗无好转。患儿为第1胎,足月自然生产,5月龄竖头,8月龄独坐,15月龄独走,自幼跑跳差,能上下楼梯、较慢,智力正常。无高血压、糖尿病、神经肌肉病家族史。1岁患肺炎,5岁患重症肺炎。
体格检查:神志清,反应好,发育正常,无特殊面容,身高117 cm,体重20.0 kg,呼吸30次/min,脉搏108次/min,双侧扁桃体Ⅱ度。心、肺、腹查体未见明显异常。步态无异常,Gower征(+),轻度翼状肩胛,脊柱前凸并脊柱强直,四肢肌张力偏低,肌力轻度下降,近端明显,为Ⅳ+,远端Ⅴ-至Ⅴ,双侧膝腱反射减弱。辅助检查:血常规:白细胞9.0×109/L,红细胞4.64×1012/L,血红蛋白125 g/L,血小板320×109/L,中性粒细胞0.51,淋巴细胞0.36。胸X线片:双肺纹理稍粗,心膈未见异常。超声心动图:轻-中度肺动脉高压,右心室肥厚。肺功能:因患儿不能配合未完成。头颅磁共振成像(MRI)未见异常;血清肌酸激酶正常;肌电图未见肌源性或神经源性损害的特征性改变;下肢肌肉MRI:双侧臀大肌重度脂肪浸润伴萎缩。当夜PSG及同步经皮CO2监测显示:患儿睡眠中无打鼾,无矛盾呼吸,呼吸事件以通气不足为主要表现,平均经皮血氧饱和度(SpO2):0.90,最低0.73;平均经皮二氧化碳(EtCO2):74 mmHg(1 mmHg=0.133 kPa),最高86 mmHg,EtCO2>50 mmHg的时间占总睡眠时间的96.2%。PSG趋势图见图1。
SpO2:经皮血氧饱和度;EtCO2:经皮二氧化碳;W:清醒期;R:快眼动睡眠期;N1:非快眼动睡眠期1期;N2:非快眼动睡眠期2期;N3:非快眼动睡眠期3期;1 mmHg=0.133 kPa
基因检查:北京康旭医学检验所进行患儿及其父母基因测序,SEPN1基因发现c.2_3insGGGCC、c.3_4insGGCCGGGCCC的杂合核苷酸变异。患儿上述变异分别遗传自父母,其父母均只携带其中一个杂合变异(图2)。
箭头为基因变异位点
采用手工压力滴定并对睡眠、呼吸及经皮CO2行同步监测。使用鼻罩,采用双水平自主/时间控制可切换模式(S/T模式),吸气压力与呼气压力分别为14和4 cmH2O (1 cmH2O=0.098 kPa),治疗开始后,患儿平均SpO2 0.94,平均EtCO2 72 mmHg (图3) 。3个月后进行呼吸机压力再次调试,吸气压力与呼气压力分别为14和5 cmH2O,平均SpO2 0.98,平均EtCO2 66 mmHg (图4)。
SpO2:经皮血氧饱和度;EtCO2:经皮二氧化碳;W:清醒期;R:快眼动睡眠期;N1:非快眼动睡眠期1期;N2:非快眼动睡眠期2期;N3:非快眼动睡眠期3期;1 mmHg=0.133 kPa
SpO2:经皮血氧饱和度;EtCO2:经皮二氧化碳;W:清醒期;R:快眼动睡眠期;N1:非快眼动睡眠期1期;N2:非快眼动睡眠期2期;N3:非快眼动睡眠期3期;1 mmHg=0.133 kPa
呼吸机治疗半年后电话随访,患儿夜间睡眠好,人机同步好,晨起无头痛、乏力。复查超声心动,肺动脉高压及右心室肥大较前好转。
本例患儿入睡后出现肺泡通气不足,经整夜PSG及同步EtCO2监测显示患儿有睡眠中呼吸运动变浅、通气不足、持续低氧血症和CO2潴留,EtCO2>50 mmHg的时间占总睡眠时间的百分比超过25%,根据国际睡眠疾病分类第三版(ICSD-3)[1]诊断为睡眠相关低通气疾病。结合患儿病史、查体及基因检查SEPN1基因变异,故确诊为SEPN1基因相关肌病。如果不予治疗,可能出现红细胞增多症、肺动脉高压、心功能不全等,因此,应早期诊断和治疗,以避免严重并发症的发生。由于神经肌肉病患者早期的呼吸障碍不是呼吸衰竭,而是夜间低通气,因此PSG已被推荐作为评估神经肌肉病患者呼吸障碍的重要方法[2]。在不明原因的肺动脉高压及右心室肥大的患者,也应注意低通气的可能。本例患儿存在肺动脉高压、右心室肥厚,PSG显示夜间入睡后SpO2持续下降,没有打鼾等阻塞性呼吸事件的表现,加之EtCO2监测结果明确了诊断。患儿在门诊诊治,未能行血气检测,由于夜间连续血气检测的不可行性,国际睡眠研究会指出,EtCO2监测可作为替代方法诊断低通气疾病;此外,因患儿呼吸肌肉无力,不能完成肺功能吸气再用力呼气的动作,未能完成肺功能检查,是本病例遗憾之处。
SEPN1基因相关肌病病程缓慢,主要表现为生长发育过程中肌张力低下、大运动发育落后、脊柱强直、活动耐力下降、呼吸功能障碍等。由于患者早期运动发育基本正常,四肢活动没有明显无力表现,进行性肌肉病变的证据有限(肌酸激酶正常或轻度升高),且肌病过程相对缓慢[3],往往容易被家长和临床医生忽视。但是,患者入睡后呼吸肌群无力、呼吸中枢敏感性下降明显,因此,睡眠相关低氧血症主要发生于睡眠期,这种夜间的血气异常可以导致患者心血管和神经认知损害,甚至猝死,值得重视。基因检查可明确SEPN1基因相关肌病的诊断。本例患儿自幼有活动耐力下降、易疲劳表现,考虑神经肌肉病,SEPN1基因检测发现c.2_3insGGGCC、c.3_4insGGCCGGGCCC的复合杂合变异。在蛋白质水平,上述变异分别导致从第5号氨基酸Arg开始的氨基酸合成发生改变,并在改变后的第63个氨基酸终止(p.Arg5GlyfsTer63)、从第8号氨基酸Gln开始的氨基酸合成发生改变,并在改变后的第78个氨基酸终止(p.Gln8ProfsTer78),均为移码变异,均可能导致蛋白质功能受到影响,均不属于多态性变化(参考数据库:1000Genomes、dbSNP)。受检者上述变异分别遗传自父母(均只携带其中一个杂合变异),符合常染色体隐性遗传病遗传方式。上述变异均尚未见文献报道(参考数据库:HGMD Pro及PubMed)。
睡眠低通气患者的呼吸异常表现为中枢性低通气,因此,需要采用双水平呼吸机的模式[4]。Annane等[4]回顾了41例SEPN1基因相关肌病的临床资料,在15岁时,50%的患儿需要无创呼吸支持,而到20岁时这一比例增加到了75%。本患儿6岁时确诊,已经出现严重的夜间低氧血症和CO2潴留,并合并肺动脉高压和右心室肥厚,表明不同疾病个体呼吸功能损害的进程不同,定期的监测、及时的干预非常重要。无创通气治疗在神经肌肉病引起的睡眠相关低通气的患儿能够成功实施,并达到满意的治疗效果,从而减缓神经肌肉病患儿疾病的进程,避免其早期即出现呼吸、循环衰竭,减少了医疗负担[5]。