成人神经元核内包涵体病的脑部MRI表现
2019年9月
中华放射学杂志,第53卷第9期 第772页-第774页
楼海燕,严志强,王小丽,刘晓燕,柯达强,李君,俞妮妮,梁辉
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)是一种少见的进展缓慢的神经元退行性疾病,病理特征是在神经元、神经胶质细胞和全身器官组织细胞中存在嗜酸性透明核内包涵体[1]。在应用皮肤活检诊断本病以前,仅依靠尸检确诊,因此曾被认为是罕见疾病。其发病机制目前尚不明确。有关NIID影像表现国内报道鲜见,临床医师对此认识不足。因此,我们将2015至2018年最终诊断为NIID的患者影像学资料进行回顾性分析,结合文献回顾分析MRI特点,提高对该病的认识。
选取浙江大学医学院附属第一医院2015年1月至2018年11月经神经内科门诊或住院收治、病理活检证实为NIID的4例患者,其中男3例,女1例。年龄分别为78、61、66和68岁。4例患者均有不同程度的长期认知功能损害。1例因突发昏迷,1例因肺部感染后意识状态下降入院后确诊,2例因头晕完善头颅MR检查后根据其特征影像学表现而被发现。高血压病史3例,糖尿病史2例,4例中有2例为兄妹关系。实验室检查提示全血细胞计数、肝肾功能指标正常。所有患者的脑脊液检查常规生化无特殊。2例患者简易智力状态检查量表(mini-mental state examination,MMSE)低于24分。另2例不能配合检查。3例患者行皮肤活检术。3例男性患者FMR1基因检测无突变。
4例患者均行头颅MRI平扫(包括扩散)或平扫+增强检查。MRI采用美国GE Signa HDXt 3 T MR扫描仪或荷兰Philips Achieva 3 T MR扫描仪,16通道头颈联合线圈。GE扫描参数:自旋回波液体衰减反转恢复(FLAIR)序列T1WI,TR 1 820 ms,TE 23 ms,TI 720 ms;T2WI,TR 4 539 ms,TE 106 ms,层厚6 mm,层间距1 mm;FOV 240 mm×240 mm。Philips扫描参数:自旋回波FLAIR序列T1WI,TR 2 000 ms,TE 20 ms,TI 800 ms;T2WI,TR2 500 ms,TE 90 ms,层厚6 mm,层间距1mm;FOV 230 mm×230 mm。DWI采用自旋回波-回波平面成像序列,TR 5 129 ms,TE 82.4 ms,激励次数1次,扩散敏感梯度用于3个正交方向,并且取2个扩散梯度因子(b值)0、1 000 s/mm2。增强T1WI,TR 12.3 ms,TE 5.1 ms,经肘静脉按照0.1 mmol/kg体质量自动推注Gd-DTPA(广州康臣药业公司),增强包括轴面、冠状面及矢状面,注射流率3 ml/s。
4例患者头颅MRI平扫显示T2WI侧脑室旁及半卵圆中心区脑白质呈弥漫对称性高信号(
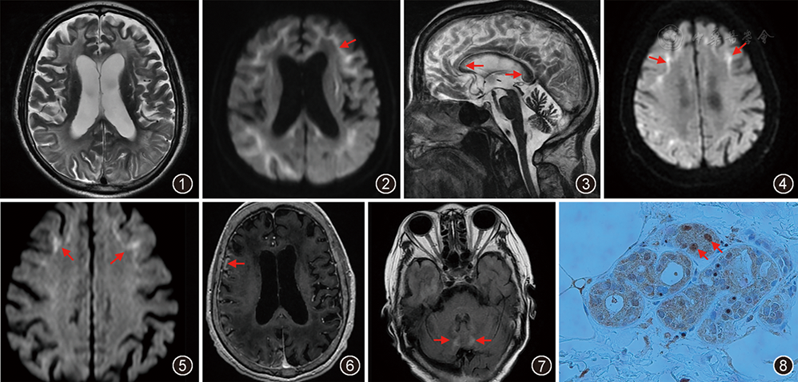
3例患者取外踝上10 cm处皮肤进行活检,术后病理提示在部分汗腺细胞、脂肪细胞和纤维细胞的核内可见嗜酸性包涵体,包涵体P62抗体、嗜酸性泛素抗体呈强阳性染色(
NIID生前诊断依靠直肠活检或腓肠神经活检。由于内镜下直肠黏膜下剥离手术难度高,且有穿孔的严重风险,而神经活检有足背皮肤麻木或感觉丧失的后遗症,因此生前能被确诊为NIID的病例非常少。随着诊断散发性[2]和家族性[3]NIID的皮肤活检手段得到广泛应用后,NIID确诊数量较前增加。由于本病可发生在婴儿至老年的任何时期,因此Takahashi-Fujigasaki[4]将其分为婴儿型、青少年型和成人型,成人型好发年龄为50~70岁[5]。40岁以上病例最主要的临床症状是痴呆,而40岁以下病例最常见的临床症状是肢体无力[6]。本组资料中有2例为兄妹关系,其中68岁的男性由于"认知功能下降"住院,其妹因"尿失禁"入院后查头颅MRI发现异常,二人均经皮肤活检后病理明确,诊断为家族性NIID。本病还可观察到脑脊液蛋白升高,运动或感觉神经传导异常,MMSE评分下降。根据Fujita等[7]报道,本病的SPECT表现与中风或癫痫有明显区别,呈现出慢性低灌注表现。
结合文献查阅,本组患者的影像表现有以下共性:(1)脑白质异常信号。T2WI可见两侧额顶叶为主的脑皮层下白质区弥漫对称性、斑片状高信号灶。无论是散发性还是家族性成人型NIID患者,所有以痴呆为主要临床表现的病例在头颅MRI中均可出现这种表现,且白质区异常信号越广泛,痴呆症状越严重。Sugiyama等[8]认为脑白质区星形胶质细胞功能障碍是其损害的可能原因。(2)DWI沿着皮髓质交界区呈曲线样高信号。本组资料显示异常高信号首先累及两侧额叶皮髓质交界,随病程进展逐渐向后方的顶叶延伸。此特点同样与患者的痴呆症状相关,也是该病经典的影像学表现。病理学改变是神经元弥漫性的髓磷脂和轴突缺失导致的白质损害[9]。无论病程长短,异常高信号仅局限在皮髓质交界,并不会累及大脑深部白质区。一般认为,该异常信号范围会随着病情进展从前向后越来越广泛,Kawarabayashi等[10]对1例NIID患者随访5年后发现DWI高信号消失而临床症状却逐渐加重。(3)幕上脑室扩大。本组病例中2例患者表现为两侧侧脑室及第三脑室扩大,患者腰椎穿刺提示颅内压正常。是否能够提示正常压力性脑积水尚待临床进一步证实,推测可能与脑萎缩有关。(4)小脑萎缩。FLAIR序列T2WI显示小脑深部白质对称性斑片状高信号。本组4例患者影像学均提示小脑萎缩,2例患者存在小脑白质区域异常高信号。Sloane等[11]关于NIID小脑病理改变的报道中,认为这种高信号与小脑神经元的脱髓鞘有关。本组病例中行走不稳、肌力下降的患者均可见上述影像表现。(5)脑膜强化或皮层脑回样强化。本组中有3例患者具有以上表现,可能与亚急性炎性发作有关,病变区域动脉扩张充血,周围脑组织水肿,注射钆对比剂后引起强化。(6)T2WI和FLAIR序列T2WI可见胼胝体压部边界清楚的高信号。
对于成人型NIID,主要需与下列疾病相鉴别:(1)脆性X相关的震颤/共济失调综合征(FXTAS),是一种罕见的遗传性神经退行性疾病,主要表现为小脑性共济失调。FMR1基因检测有助于二者鉴别。FXTAS典型MRI特点为FLAIR、T2WI小脑中脚对称性高信号病灶和全小脑萎缩,侧脑室周围深部脑白质病变,可出现胼胝体压部高信号病灶。而NIID患者DWI特征的皮髓质交界区曲线状高信号可能有一定的鉴别意义。(2)Binswanger病,又名皮质下动脉硬化性脑病,MRI特点为不同程度脑萎缩及脑室扩大,在侧脑室周围深部脑白质对称性分布月晕状长T1、长T2信号,常见基底节、丘脑及脑干梗死。而NIID患者几乎没有基底节、丘脑和半卵圆中心区的腔隙性脑梗死灶。(3)MELAS综合征,又名线粒体脑肌病,病变多分布在皮质和皮层下白质,深部白质不受累。MRI表现为颞顶叶皮层区T2WI、DWI高信号,临床发作类似卒中。NIID病变几乎不累及皮层病变,DWI的信号变化也与MELAS有较大差异,其白质的特异性信号异常不会出现在脑室周围白质。
综上所述,尽管成人型NIID临床表现复杂多变,但DWI皮髓质交界区呈曲线样分布的高信号仍为其特征性影像表现。对于临床高度怀疑患者需行头颅MRI检查,并结合皮肤活检和基因检测,可以提高诊断准确性。
[1] Sung JH, Ramirez-Lassepas M, Mastri AR, et al. An unusual degenerative disorder of neurons associated with a novel intranuclear hyaline inclusion (neuronal intranuclear hyaline inclusion disease). A clinicopathological study of a case[J]. J Neuropathol Exp Neurol, 1980,39(2):107-130.
[2] Sone J, Kitagawa N, Sugawara E, et al. Neuronal intranuclear inclusion disease cases with leukoencephalopathy diagnosed via skin biopsy[J]. J Neurol Neurosurg Psychiatry, 2014,85(3):354-356.
[3] Sone J, Tanaka F, Koike H, et al. Skin biopsy is useful for the antemortem diagnosis of neuronal intranuclear inclusion disease[J]. Neurology, 2011,76(16):1372-1376.
[4] Takahashi-Fujigasaki J. Neuronal intranuclear hyaline inclusion disease[J]. Neuropathology, 2003,23(4):351-359.
[5] Takumida H, Yakabe M, Mori H, et al. Case of a 78-year-old woman with a neuronal intranuclear inclusion disease[J]. Geriatr Gerontol Int, 2017,17(12):2623-2625.
[6] Sone J, Mori K, Inagaki T, et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease[J]. Brain, 2016,139(
[7] Fujita K, Osaki Y, Miyamoto R, et al. Neurologic attack and dynamic perfusion abnormality in neuronal intranuclear inclusion disease[J]. Neurol Clin Pract, 2017, 7(6):e39-42.
[8] Sugiyama A, Sato N, Kimura Y, et al. MR imaging features of the cerebellum in adult-onset neuronal intranuclear inclusion disease: 8 cases[J]. AJNR Am J Neuroradiol, 2017,38(11):2100-2104.
[9] Yokoi S, Yasui K, Hasegawa Y, et al. Pathological background of subcortical hyperintensities on diffusion-weighted images in a case of neuronal intranuclear inclusion disease[J]. Clin Neuropathol, 2016,35(6):375-380.
[10] Kawarabayashi T, Nakamura T, Seino Y, et al. Disappearance of MRI imaging signals in a patient with neuronal intranuclear inclusion disease[J]. J Neurol Sci, 2018,388:1-3.
[11] Sloane AE, Becker LE, Ang LC, et al. Neuronal intranuclear hyaline inclusion disease with progressive cerebellar ataxia[J]. Pediatr Neurol, 1994,10(1):61-66.
