参考文献
1 国家卫生计生委合理用药专家委员会.全国细菌耐药监测网.2018年全国细菌耐药监测报告(简要版)[R]. (2019-11-19)[2019-12-11].http://www.carss.cn/Report/Details/648.
2 Nordmann P, Naas T, Poirel L. Global spread of carbapenemase-producing enterobacteriaceae[J]. Emerg Infect Dis, 2011, 17(10):1791-1798. DOI: 10.3201/eid1710.110655.
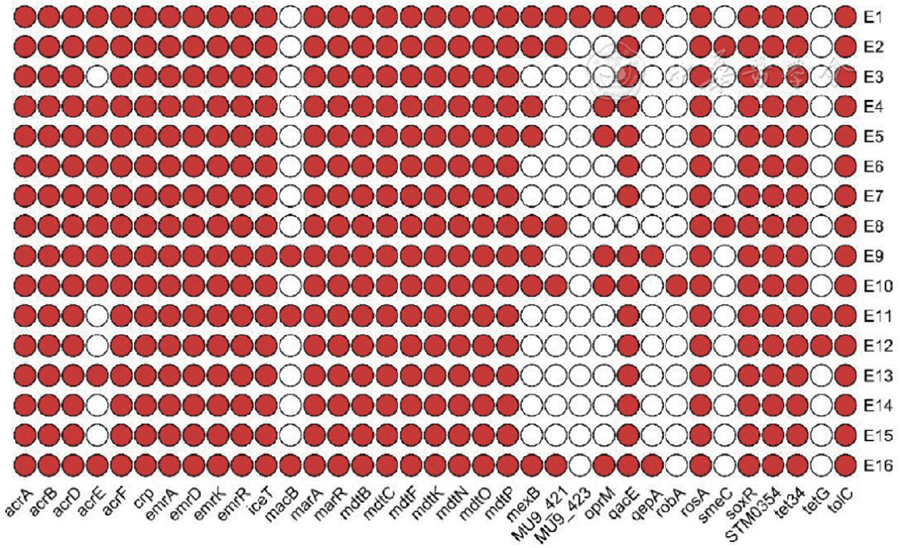
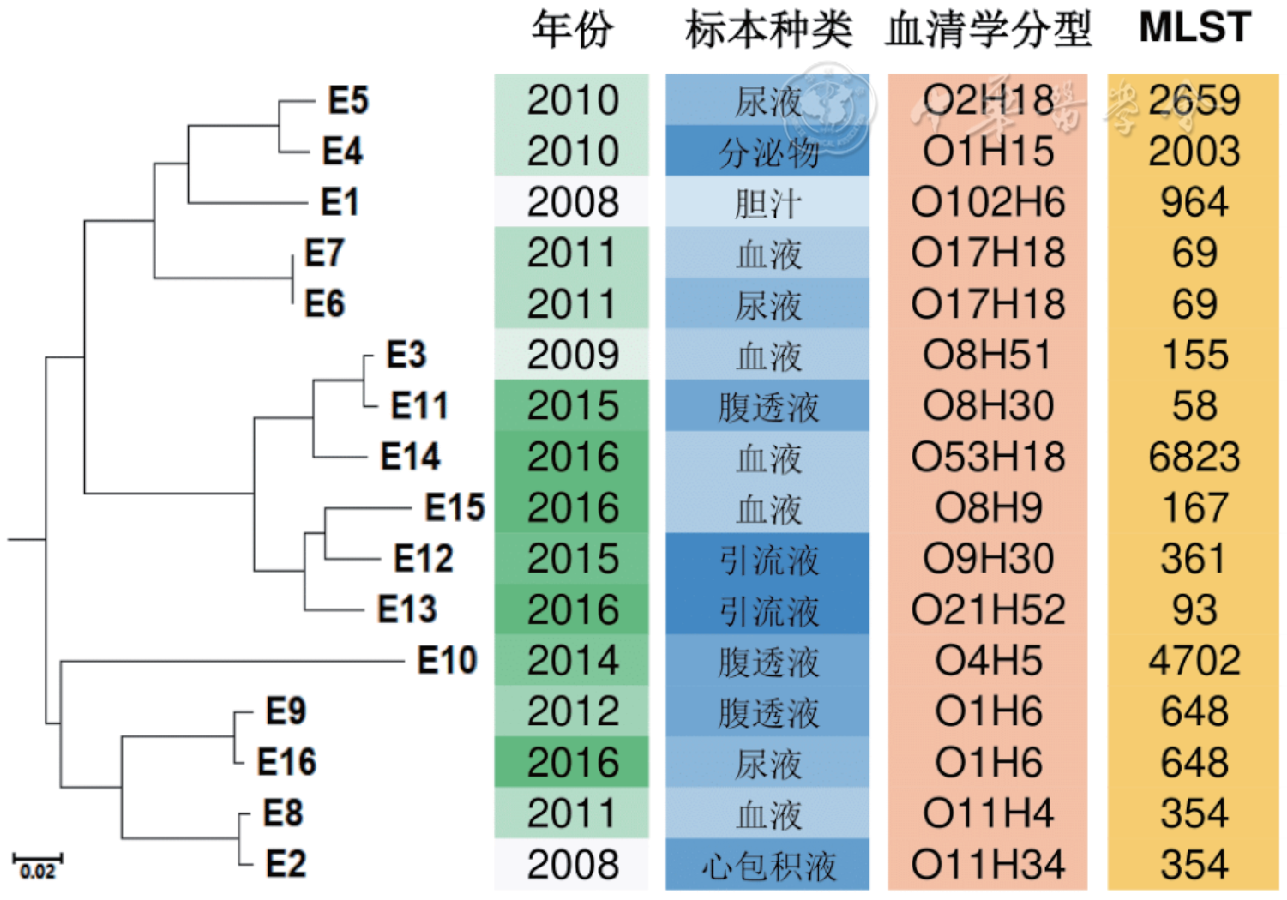
3 陈韩, 郑周, 陈思思, 等. 血流感染产碳青霉烯酶大肠埃希菌的耐药基因及同源性研究[J].中华医院感染学杂志, 2019, 29(7):971-975.
4 Zhang Y, Wang Q, Yin Y, et al. Epidemiology of carbapenem-resistant enterobacteriaceae infections: report from the China CRE network[J]. Antimicrob Agents Chemother, 2018, 62(2):e01882-17.DOI: 10.1128/AAC.01882-17.
5 Mahalingam N, Manivannan B, Khamari B, et al. Detection of antibiotic resistance determinants and their transmissibility among clinically isolated carbapenem-resistant escherichia coli from South India[J]. Med Princ Pract, 2018, 27(5):428-435. DOI: 10.1159/000489885.
6 Dagher C, Salloum T, Alousi S, et al. Molecular characterization of Carbapenem resistant Escherichia coli recovered from a tertiary hospital in Lebanon[J]. PLoS One, 2018, 13(9):e0203323. DOI: 10.1371/journal.pone.0203323.
7 国家卫生健康委员会编.中国抗菌药物管理和细菌耐药现状报告(2019)[M].北京:中国协和医科大学出版社,2019.
8 Mammeri H, Guillon H, Eb F, et al. Phenotypic and biochemical comparison of the carbapenem-hydrolyzing activities of five plasmid-borne AmpC β-lactamases[J]. Antimicrob Agents Chemother, 2010, 54(11):4556-4560. DOI:10.1128/AAC.01762-09.
9 Yamane K, Wachino J, Suzuki S, et al. Plasmid-mediated qepA gene among Escherichia coli clinical isolates from Japan[J]. Antimicrob Agents Chemother, 2008, 52(4):1564-1566. DOI: 10.1128/AAC.01137-07.
10 Fernando DM, Kumar A. Resistance-nodulation-division multidrug efflux pumps in gram-negative bacteria: role in virulence[J]. Antibiotics (Basel), 2013, 2(1):163-181. DOI: 10.3390/antibiotics2010163.
11 Nikaido H, Pagès JM. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria[J]. FEMS Microbiol Rev, 2012, 36(2):340-363. DOI: 10.1111/j.1574-6976.2011.00290.x.
12 Salipante SJ, SenGupta DJ, Cummings LA, et al. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology[J]. J Clin Microbiol, 2015, 53(4):1072-1079. DOI: 10.1128/JCM.03385-14.
13 Orlek A, Stoesser N, Anjum MF, et al. Plasmid classification in an era of whole-genome sequencing: application in studies of antibiotic resistance epidemiology[J]. Front Microbiol, 2017, 8:182. DOI: 10.3389/fmicb.2017.00182.
14 Thomsen MC, Ahrenfeldt J, Cisneros JL, et al. A bacterial analysis platform: an integrated system for analysing bacterial whole genome sequencing data for clinical diagnostics and surveillance[J]. PLoS One, 2016, 11(6):e0157718. DOI: 10.1371/journal.pone.0157718.