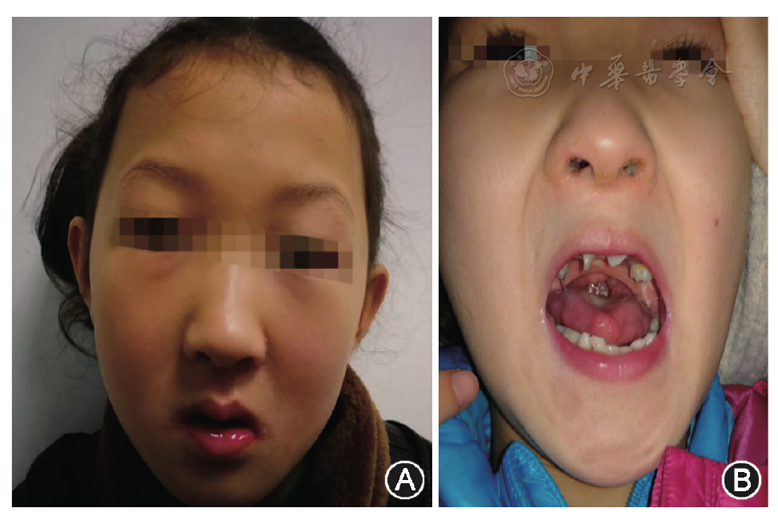
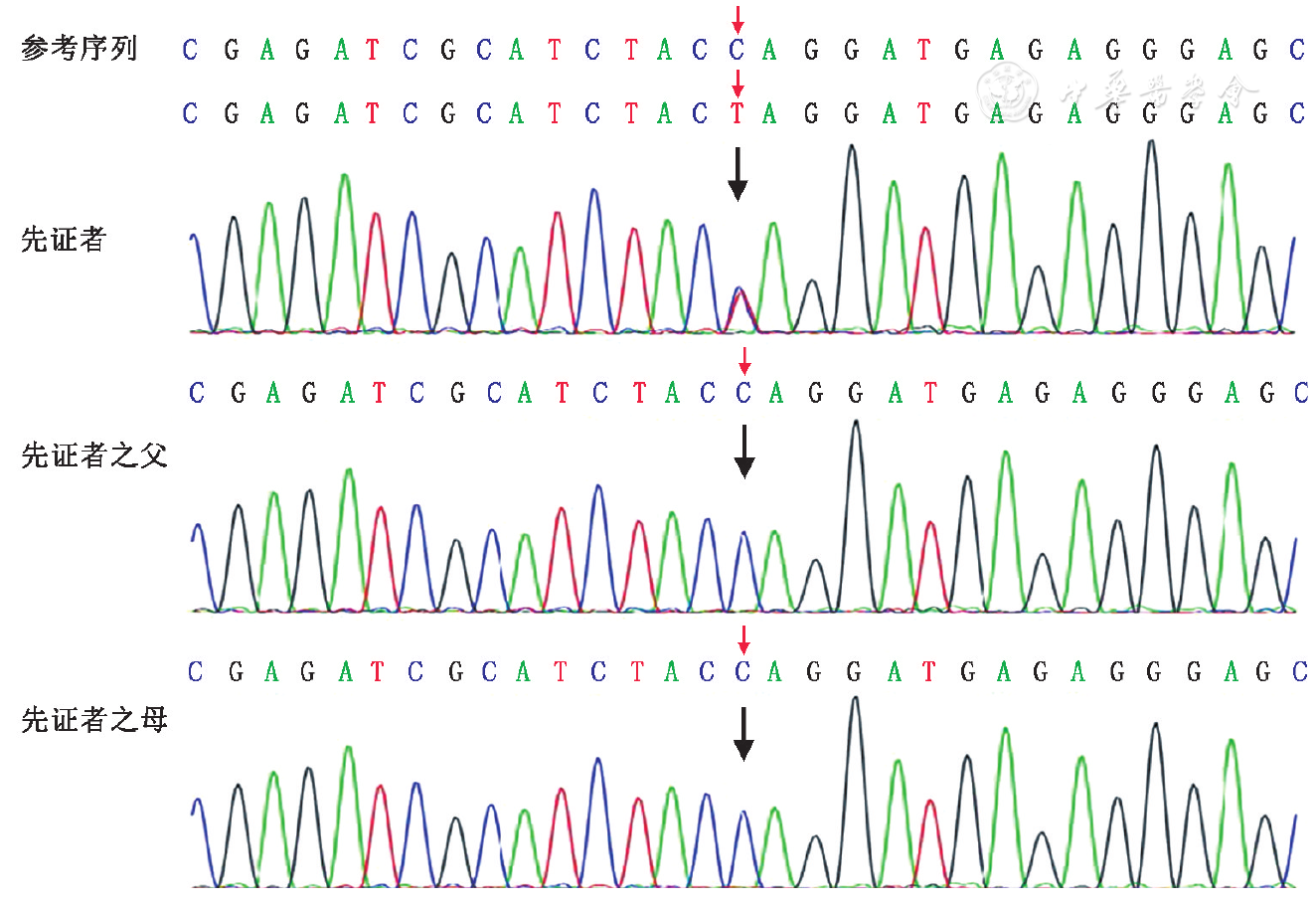
Conflicts of interest:志谢参考文献
[1] Glass IA, Swindlehurst CA, Aitken DA, et al. Interstitial deletion of the long arm of chromosome 2 with normal levels of isocitrate dehydrogenase[J]. J Med Genet, 1989, 26(2): 127-130. DOI: 10.1136/jmg.26.2.127.
[2] Docker D, Schubach M, Menzel M, et al. Further delineation of the SATB2 phenotype[J]. Eur J Hum Genet, 2014, 22(8): 1034-1039. DOI: 10.1038/ejhg.2013.280.
[3] Zarate YA, Perry H, Ben-Omran T, et al. Further supporting evidence for the SATB2-associated syndrome found through whole exome sequencing[J]. Am J Med Genet A, 2015, 167A(5): 1026-1032. DOI: 10.1002/ajmg.a.36849.
[4] FitzPatrick DR, Carr IM, McLaren L, et al. Identification of SATB2 as the cleft palate gene on 2q32-q33[J]. Hum Mol Genet, 2003, 12(19): 2491-1501. DOI: 10.1093/hmg/ddg248.
[5] Dobreva G, Dambacher J, Grosschedl R. SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression[J]. Genes Dev, 2003, 17(24): 3048-3061. DOI: 10.1101/gad.1153003.
[6] Gyorgy AB, Szemes M, de Juan Romero C, et al. SATB2 interacts with chromatin-remodeling molecules in differentiating cortical neurons[J]. Eur J Neurosci, 2008, 27(4): 865-873. DOI: 10.1111/j.1460-9568.2008.06061.x.
[7] 靳春雷,雷永良,刘姣,等. 2q33.1微缺失致SATB2基因部分缺失兄妹的表型及遗传学分析[J].中华医学遗传学杂志, 2019, 36(6): 628-631. DOI: 10.3760/cma.j.issn.1003-9406.2019.06.025.Jin CL, Lei YL, Liu J, et al. Phenotypic and genetic analysis of a sibpair with partial deletion of SATB2 gene caused by 2q33.1 microdeletion[J]. Chin J Med Genet, 2019, 36(6): 628-631. DOI: 10.3760/cma.j.issn.1003-9406.2019.06.025.
[8] Kikuno R, Nagase T, Ishikawa K, et al. Prediction of the coding sequences of unidentified human genes. XIV. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro[J]. DNA Res, 1999, 6(3): 197-205. DOI: 10.1093/dnares/6.3.197.
[9] Jugessur A, Shi M, Gjessing HK, et al. Maternal genes and facial clefts in offspring: a comprehensive search for genetic associations in two population-based cleft studies from Scandinavia[J]. PLoS One, 2010, 5(7): e11493. DOI: 10.1371/journal.pone.0011493.
[10] Asadollahi R, Oneda B, Joset P, et al. The clinical significance of small copy number variants in neurodevelopmental disorders[J]. J Med Genet, 2014, 51(10): 677-688. DOI: 10.1136/jmedgenet-2014-102588.
[11] Kaiser AS, Maas B, Wolff A, et al. Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia[J]. Eur J Hum Genet, 2015, 23(5): 704-707. DOI: 10.1038/ejhg.2014.163.
[12] Kikuiri T, Mishima H, Imura H, et al. Patients with SATB2-associated syndrome exhibiting multiple odontomas[J]. Am J Med Genet A, 2018, 176(12): 2614-2622. DOI: 10.1002/ajmg.a.40670.
[13] Gregoric Kumperscak H, Krgovic D, Vokac NK. Specific behavioural phenotype and secondary cognitive decline as a result of an 8.6 Mb deletion of 2q32.2q33.1[J]. J Int Med Res, 2016, 44(2): 395-402. DOI: 10.1177/0300060515595651.
[14] Zarate YA, Kalsner L, Basinger A, et al. Genotype and phenotype in 12 additional individuals with SATB2-associated syndrome[J]. Clin Genet, 2017, 92(4): 423-429. DOI: 10.1111/cge.12982.
[15] Scott J, Adams C, Simmons K, et al. Dental radiographic findings in 18 individuals with SATB2-associated syndrome[J]. Clinical Oral Investig, 2018, 22(8): 2947-2951. DOI: 10.1007/s00784-018-2702-9.
[16] Boone PM, Chan YM, Hunter JV, et al. Increased bone turnover, osteoporosis, progressive tibial bowing, fractures, and scoliosis in a patient with a final-exon SATB2 frameshift mutation[J]. Am J Med Genet A, 2016, 170(11): 3028-3032. DOI: 10.1002/ajmg.a.37847.
[17] Rosenfeld JA, Ballif BC, Lucas A, et al. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome[J]. PLoS One, 2009, 4(8): e6568. DOI: 10.1371/journal.pone.0006568.
[18] Lee JS, Yoo Y, Lim BC, et al. SATB2-associated syndrome presenting with Rett-like phenotypes[J]. Clin Genet, 2016, 89(6): 728-732. DOI: 10.1111/cge.12698.
[19] Britanova O, Depew MJ, Schwark M, et al. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development[J]. Am J Hum Genet, 2006, 79(4): 668-678. DOI: 10.1086/508214.
[20] Dobreva G, Chahrour M, Dautzenberg M, et al. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation[J]. Cell, 2006, 125(5): 971-986. DOI: 10.1016/j.cell.2006.05.012.
[21] Wu L, Chen J, Qin Y, et al. SATB2 suppresses gastric cancer cell proliferation and migration[J]. Tumour Biol, 2016, 37(4): 4597-4602. DOI: 10.1007/s13277-015-4282-5.
[22] Uhlén M, Fagerberg L, Hallstr?m BM, et al. Proteomics. Tissue-based map of the human proteome[J]. Science, 2015, 347(6220): 1260419. DOI: 10.1126/science.1260419.
[23] Britanova O, de Juan Romero C, Cheung A, et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex[J]. Neuron, 2008, 57(3): 378-392. DOI: 10.1016/j.neuron.2007.12.028.
[24] Alcamo EA, Chirivella L, Dautzenberg M, et al. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex[J]. Neuron, 2008, 57(3): 364-377. DOI: 10.1016/j.neuron.2007.12.012.
[25] Fish JL. Developmental mechanisms underlying variation in craniofacial disease and evolution[J]. Dev Biol, 2016, 415(2): 188-197. DOI: 10.1016/j.ydbio.2015.12.019.
[26] Rosenfeld JA, Ballif BC, Lucas A, et al. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome[J]. PLoS One, 2009, 4(8): e6568. DOI: 10.1371/journal.pone.0006568.
[27] Leoyklang P, Suphapeetiporn K, Srichomthong C, et al. Disorders with similar clinical phenotypes reveal underlying genetic interaction: SATB2 acts as an activator of the UPF3B gene[J]. Hum Genet, 2013, 132(12): 1383-1393. DOI: 10.1007/s00439-013-1345-9.