运动神经元病的诊断和分类
2019年12月
中华神经科杂志,第52卷第12期 第1065页-第1067页
樊东升,陈璐
运动神经元病(motor neuron disease,MND),是一类散发或遗传的致死性神经系统退行性疾病。它选择性累及脑和脊髓的上、下运动神经元,临床表现为进行性的肌肉萎缩、无力伴有锥体束征,言语不清、饮水呛咳,当呼吸肌受累时可出现呼吸困难、呼吸衰竭[1,2,3]。本病呈全球性分布,年发病率为(0.4~1.76)/10万,患病率为(4~6)/10万,年死亡率为2/10万。男性发病率约为女性的2倍,多数患者起病年龄大于45岁,且每隔20年发病率逐渐增加。
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)是MND中最常见的一种形式,也是这类神经系统变性疾病中最具破坏性的一种。目前,部分学者倾向于使用ALS代指MND[4]。这里,我们以MND作为统称概念。
MND起病隐匿,首发症状常为单个肢体的无力或言语不清,不易引起患者重视,且容易与颈椎病、周围神经病等疾病相混淆[5,6,7]。但是尽早诊断MND有助于患者在运动神经元存活较多时就开始神经保护治疗,并能够帮助患者及其家庭做好未来规划,且有利于患者的情绪调整[5,6]。遗憾的是,目前MND尚无特征性的生物标志物,诊断主要依据患者的临床表现和肌电图检查结果;脑脊液检查、脊柱核磁等有助于排除其他疾病,但无法确诊MND[8]。
脑干和脊髓各节段的上、下运动神经元受累体征是诊断MND的重要依据,而肌电图是判断亚临床下运动神经元受累的重要辅助检查。MND的诊断需要依赖医师的临床经验和判断,经验不足容易造成疾病的误诊和诊断延迟,而易于理解且便于实施的诊断标准能够提高临床医师诊断的准确性,并缩短诊断延迟时间。
目前国际上关于ALS的研究最为深入、诊断标准最为完善。迄今为止,国际公认的ALS诊断标准共有3种,按时间先后顺序依次为El Escorial诊断标准[9]、Airlie House诊断标准(又称修订版El Escorial诊断标准)[10]和Awaji-shima电生理诊断标准[11]。1994年,世界神经病学联盟(WFN)首次发布了El Escorial诊断标准,它基于患者的临床表现及疾病累及范围,将ALS分为确诊(definite)、拟诊(probable)、可能(possible)及疑诊(suspected)4个不同等级。El Escorial诊断标准为ALS的诊断提供了简单实用的方法,自此诊断标准提出后,ALS的诊断有了统一的依据,极大地提高了临床诊断的效率和准确性[9]。
随后,由于神经电生理技术的发展,为进一步提高诊断的敏感性与准确性,WFN于1998年重新修订了El Escorial诊断标准,将肌电图作为检测下运动神经元损害的重要手段,在确诊、拟诊、可能3个等级的基础上,又基于患者的肌电图表现,引入了实验室支持拟诊级ALS的概念(

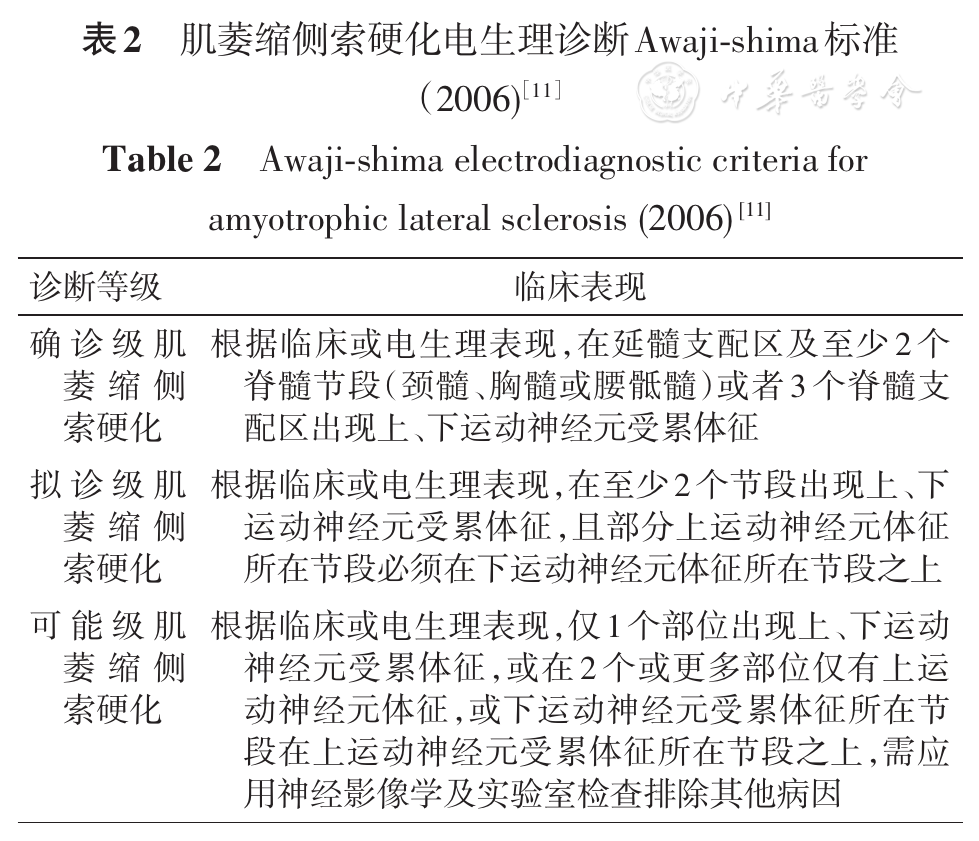
随着对ALS认识的进一步深入和神经电生理检查的广泛应用,在上述诊断标准的基础上,2006年又在日本制定了Awaji-shima电生理诊断标准。该标准认为临床症状、体征与肌电图表现在诊断下运动神经元损害方面具有同等重要的意义,因而又取消了实验室支持拟诊ALS这一等级,将ALS诊断级别仅分为确诊、拟诊、可能3个等级(
MND存在多种分类方法,根据病因可将其分为2个临床类型:(1)家族性,约占5%~10%,其中国内患者最常见的致病基因为SOD1基因突变,欧美白人最常见的致病基因则为C9ORF72基因;(2)散发性,约占90%以上,病因不明。
临床上,最常见的是按照起病部位及临床表现的不同,将其分为如下类别:
上肢或下肢首先出现上、下运动神经元受累体征,此型占患者总数的70%。
以言语不清和吞咽困难为首要表现,随后出现肢体受累症状,此型占患者总数的25%。
此型较为少见,表现为40岁以后起病,4年内仅有上运动神经元受累而不出现下运动神经元受累,4年内出现下运动神经元受累表现者诊断为以上运动神经元受累为主要表现的ALS。
此型仅有下运动神经元受累体征,并具有明显的临床异质性。
如连枷臂综合征(FAS)和连枷腿综合征(FLS),FAS和FLS均表现为症状和体征局限于肢体1个区域达12个月以上而不出现其他区域受累的体征[8,12,13,14,15,16]。
但是临床研究发现,多数PLS和PMA的患者随病情逐渐进展最终会出现上、下运动神经元同时受累,此时其临床表现与ALS相同,因此部分学者认为PLS和PMA是ALS中较为特殊的类型[12]。无论哪一种MND的临床表型,随疾病发展,最后终将累及呼吸肌,造成呼吸困难,直至呼吸衰竭;大多数MND患者因呼吸衰竭或营养不良去世[12,13,14,15,16]。
综上所述,MND是一种成人起病的致死性神经系统退行性疾病,可分为肢体起病型ALS、球部起病型ALS、PMA、PLS、FAS、FLS等多种类型,其中绝大部分患者可按照修订版El Escorial诊断标准或Awaji-shima诊断标准进行诊断。目前本病尚无有效逆转疾病的方法,但包括营养支持、呼吸管理及药物治疗在内的综合治疗,可以在一定程度上延缓疾病进程,延长患者的生存时间并提高患者生活质量。
[1] Chiò A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: A critical review[J]. Amyotroph Lateral Scler, 2009, 10(5-6): 310-323.
[2] Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis[J]. Nat Rev Neurosci, 2013, 14(4): 248-264.
[3] Mandrioli J, Faglioni P, Nichelli P, et al. Amyotrophic lateral sclerosis: prognostic indicators of survival[J]. Amyotroph Lateral Scler, 2006, 7(4): 211-220.
[4] 蒋雨平.运动神经元病[J].中国临床神经科学, 2014, 22(6):
[5] EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis, Andersen PM, Abrahams S, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)-revised report of an EFNS task force[J]. Eur J Neurol, 2012, 19(3): 360-375.
[6] 樊东升,张俊,邓敏,等.肌萎缩侧索硬化/运动神经元病的基础与临床研究[J].北京大学学报(医学版), 2009, 41(3): 279-281.
[7] 中华医学会神经病学分会肌电图与临床神经电生理学组,中华医学会神经病学分会神经肌肉病学组.中国肌萎缩侧索硬化诊断和治疗指南[J].中华神经科杂志, 2012, 45(7):531-533.
[8] 卢家红.易与运动神经元病混淆的疾病[J].中国实用内科杂志, 2009, 29(2): 105-107.
[9] Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on motor neuron diseases/amyotrophic lateral sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors[J]. J Neurol Sci, 1994, 124
[10] Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis[J]. Amyotroph Lateral Scler Other Motor Neuron Disord, 2000, 1(5): 293-299.
[11] de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS[J]. Clin Neurophysiol, 2008, 119(3): 497-503.
[12] Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis[J]. Lancet, 2011,377:942-955.
[13] de Carvalho M. Natural history of young-adult amyotrophic lateral sclerosis[J]. Neurology, 2009, 73(8):
[14] Singer MA, Statland JM, Wolfe GI, et al. Primary lateral sclerosis[J]. Muscle Nerve, 2007, 35: 291-302.
[15] Wijesekera LC, Mathers S, Talman P, et al. Natural history and clinical features of the flail arm and flail leg ALS variants[J]. Neurology, 2009, 72(12): 1087-1094.
[16] Couratier P, Truong C, Khalil M, et al. Clinical features of flail arm syndrome[J]. Muscle Nerve, 2000, 23(4): 646-648.
单选题(授予Ⅱ类学分说明及答题二维码见杂志内活插页)1.下列哪一项不属于肌萎缩侧索硬化的诊断标准?A. Airlie House诊断标准B. Awaji-shima诊断标准C. El Escorial诊断标准D. McDonald诊断标准2.下列哪一项不是运动神经元病相关的临床表现?A.肌肉萎缩无力B.晨轻暮重C.言语不清、饮水呛咳D.呼吸困难3.下列哪一项是Airlie House诊断标准特有的诊断级别?A.确诊级B.拟诊级C.实验室支持拟诊级D.可能级4.下列哪一种综合征属于运动神经元病的范畴?A.连枷臂综合征B.脊髓半切综合征C.脊髓灰质炎后综合征D.肌无力综合征5.男性,50岁,主因进行性四肢无力1.5年入院,目前双上肢抬举困难、行走费力,伴言语不清,无憋气、活动后气短。体检:舌肌可见萎缩及纤颤,双侧掌颌反射、下颌反射、吸吮反射阳性,四肢肌肉萎缩、力弱,四肢腱反射活跃,双侧霍夫曼征、巴宾斯基征阳性。感觉系统体检无异常。神经传导检查未见明显异常。肌电图提示:双侧胸锁乳突肌、双侧第一骨间肌、双侧腹直肌、双侧胫前肌神经源性损害。按照Airlie House诊断标准,该患者最为可能的分级是?A.可能级B.拟诊级C.确诊级D.实验室支持拟诊级