Noonan综合征是一类常染色体显性遗传性疾病,伴有多器官系统受累,主要临床特征为特殊面容、先天性心脏病、心肌病变、身材矮小、发育迟缓、肾脏畸形、凝血功能障碍等。发病率约为1/2 500~1/1 000,在合并心脏病变的各种综合征中,Noonan综合征是第二常见综合征,仅次于21三体综合征。肺动脉狭窄为Noonan综合征最常见的心脏合并症,发生率高达50%~60%,其次为肥厚型心肌病(20%)及房间隔缺损(6%~10%)[1]。基因检测可以协助本病的诊断,并提供相关遗传咨询及指导临床治疗。本研究分析5例Noonan综合征合并肥厚型心肌病患儿的基因突变特点及临床表现,以期从遗传分子生物学角度进一步认识Noonan综合征不同基因突变与肥厚型心肌病的关系及临床意义。
以北京安贞医院小儿心脏中心2014—2016年门诊确诊的5例Noonan综合征合并肥厚型心肌病患儿为研究对象。5例均为散发病例,无家族聚集性。患儿特殊面容和临床特征符合Noonan综合征的诊断标准[2]。
5例患儿均在北京安贞医院小儿心脏中心由专业医生进行超声心动图诊断和评估。超声诊断仪采用Philips Epiq 7C和APLIO ARTIDA SSH-880CV高端心脏诊断系统,探头频率5~8 MHz。检查内容包括:心脏结构,心腔及大血管内径,心室壁厚度,各瓣膜功能及血流,压力阶差,左、右心室流出道血流及压力阶差,左、右心室收缩功能。超声测量方法依据美国超声心动图学会儿童测量写作组(Pediatric Measurements writing group of ASE),儿童及先天性心脏病委员会(Pediatric and Congenital Heart Disease Council)2010年发布的《小儿超声心动图定量测量方法建议》[3]。
基因全外显子测序由北京信诺佰世医学检验所和北京智因东方转化医学研究中心协助完成。从受检者外周血中提取基因组DNA,构建基因组文库,通过探针杂交捕获与Noonan综合征相关基因外显子及相邻内含子区域(50 bp),并进行富集。富集的目的基因片段通过下代高通量测序仪(Illumina)进行测序。测序数据运用NextGene V2.3.4软件与UCSC数据库提供的人类基因组hg19参考序列进行比对,并对目标区域的覆盖度和测序质量进行评估。依据严格的筛选标准对变异进行过滤,并添加HGMD数据库、蛋白功能预测软件的相关注释信息。对明确或可能与受检者临床表型相关的基因变异,采用Sanger测序进行验证。对本组检出的3个Noonan综合征相关基因分析突变基因特征,确定突变位点的结构域。
截至2017年5月1日在Pubmed数据库中分别检索关键词"PTPN11""RAF1""RITI""Hypertrophic Cardiomyopathy",共检索出140篇相关文献,分析其中关于本研究所涉及的突变位点与肥厚型心肌病的关系,记录与报道的突变位点位于同一结构域的病例,同时对比文献报道与本研究中患儿心肌肥厚及心脏结构异常的特点。
5例患儿中2例患儿基因检测发现与肥厚型心肌病相关的已知基因突变MYPN、MYH6及MYBPC3。2例患儿基因突变均为RAF1,c.770C>T,但临床表现不同(表1)。
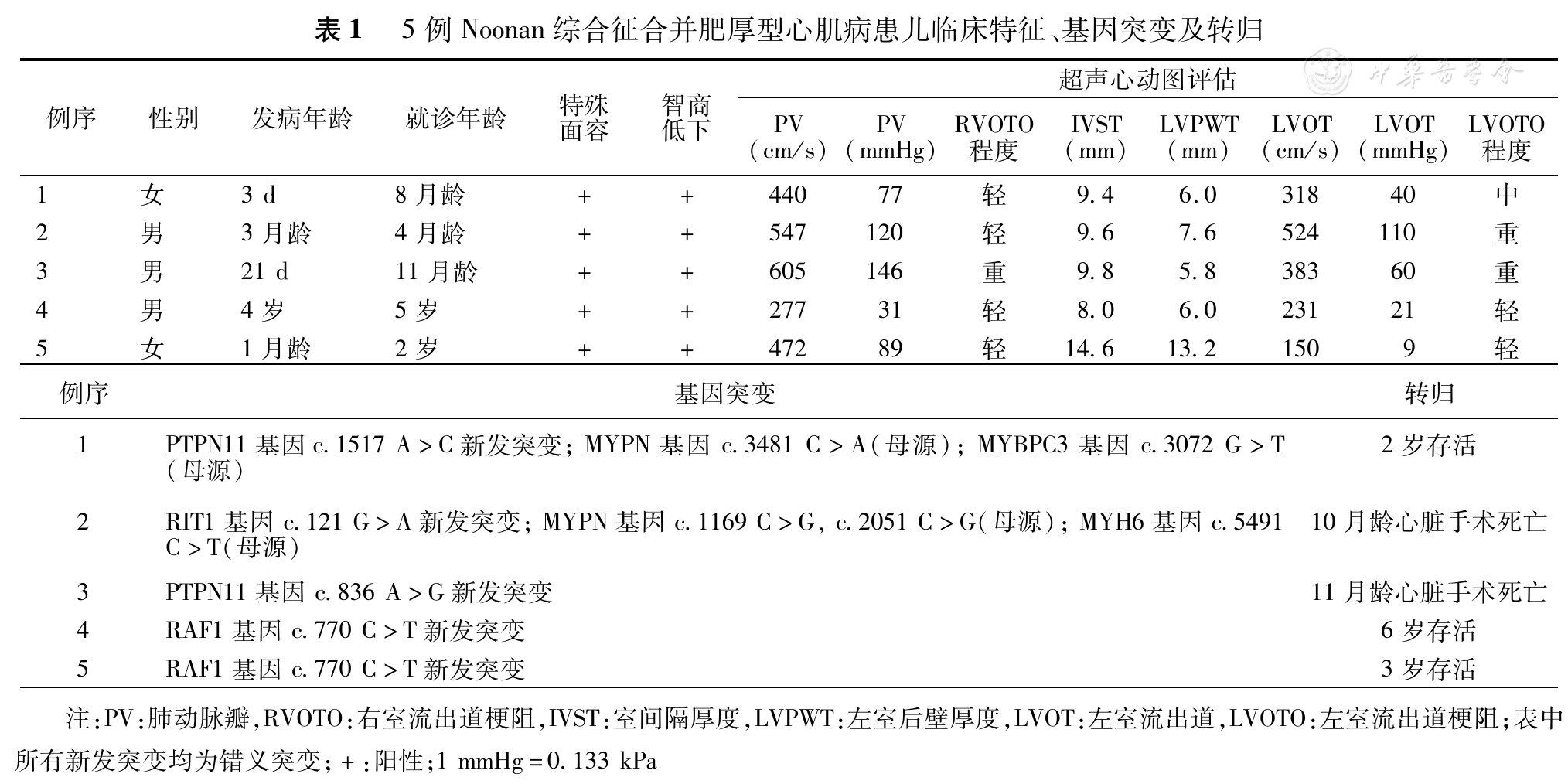
例1 女,8月龄,特殊面容,眼裂小,眼距宽,鼻唇沟浅,无明显喂养困难,智商低于同龄儿童。超声心动图示:左室壁心肌增厚,室间隔厚度9.4 mm,左室后壁6 mm,左室舒张末径20 mm,收缩末径11 mm,收缩期心尖部近闭塞,射血分数0.81,左室流出道流速增快318 cm/s;压差40 mmHg(1 mmHg=0.133 kPa);右室流出道狭窄,内径8 mm,肺动脉瓣重度狭窄,瓣口最大流速440 cm/s,跨瓣压差77 mmHg,卵圆孔未闭3 mm。生后染色体核型分析未见异常。基因突变检测结果:患儿12号染色体PTPN11基因的第13外显子区域检到一杂合的c.1517A>C碱基取代,该突变为新发突变,导致该蛋白第506位的谷氨酰胺改变为脯氨酸(p.506,Q>P)。基因检测同时发现来自母源的MYPN基因(10号染色体,c.3481C>A,p.1161L>I)和MYBPC3基因(11号染色体,c.3072G>T,p.1024S>R)杂合突变。母亲无相关临床症状及肥厚型心肌病家族史。
例2 男,4月龄,特殊面容,眼距宽,鼻头大,耳位低,有颈蹼,生长发育稍落后。超声心动图示:双侧室壁肥厚,间隔最厚处9.6 mm,左室后壁7.6 mm,左室舒张末径20 mm,收缩末径8.6 mm,收缩期中下段心腔近闭塞,射血分数0.90;左室流出道梗阻明显,最大流速524 cm/s,压差110 mmHg,收缩期前向运动(SAM)征(+)。右室壁增厚,厚度5.2 mm,流出道内径尚可,肺动脉瓣重度狭窄,瓣上最大流速547 cm/s,跨瓣压差120 mmHg ,卵圆孔未闭3 mm。基因突变检测结果:患儿1号染色体RIT1基因的第3外显子区域检到一杂合的c.121G>A碱基取代,该突变为新发突变,导致该蛋白第41位的丙氨酸改变为苏氨酸(p.41,A>T)。同时发现来自母源的MYPN基因(10号染色体,c.1169C>G,p.390S>C; c.2051C>G, p.684,S>C)和MYH6基因(14号染色体,c.5491G>A,p.1831E>K)杂合突变。母亲无相关临床症状及肥厚型心肌病家族史。
例3 男,11月龄,特殊面容,方颅,眼距宽,耳位低,新生儿期曾诊断先天性心脏病:肺动脉瓣重度狭窄,动脉导管未闭,卵圆孔未闭。超声心动图示:双侧心室壁及室间隔高度肥厚,室间隔最厚处9.8 mm,左室后壁5.8 mm。左室舒张末径20 mm,收缩末径11 mm,射血分数0.80,左、右心室流出道收缩期内径明显狭窄,流速加快,左室流出道收缩期最大流速388 cm/s,压差60 mmHg,SAM征(+),右室前壁增厚,厚度6 mm,右室流出道重度狭窄,收缩期4.3 mm,肺动脉瓣重度狭窄,肺动脉瓣上流速605 cm/s,压差146 mmHg。卵圆孔未闭3 mm,心包积液少量。基因突变检测结果:患儿12号染色体PTPN11基因的第7外显子区域检到一杂合的c.836A>G碱基取代,该突变为新发突变,导致该蛋白第279位的酪氨酸改变为半胱氨酸(p.279,Y>C)。未检出与肥厚型心肌病相关的其他已知基因突变。
例4 男,5岁,特殊面容,头颅比例大,眼距宽,眼角下斜,眉下斜,发际低,有颈蹼,耳位低,隐睾,生长及运动发育可,语言水平落后,对答尚可。超声心动图示:室间隔增厚,最厚处8 mm,左室后壁厚度6 mm,左室舒张末径26 mm,收缩末径13 mm,左室射血分数0.80,左室流出道最大流速231 cm/s,压差21 mmHg;肺动脉瓣轻度狭窄,肺动脉瓣上流速277 cm/s,压差31 mmHg,距肺动脉瓣下12 mm处见肥厚肌束致管腔轻度狭窄,房间隔缺损(Ⅱ孔型) 10 mm。基因突变检测结果:患儿3号染色体RAF1基因的第6外显子区域检到一杂合的c.770C>T碱基取代,该突变为新发突变,导致该蛋白第257位的丝氨酸改变为亮氨酸(p.257,S>L)。未检出与肥厚型心肌病相关的其他已知基因突变。
例5 女,2.5岁,特殊面容,方颅,眼距宽,耳位低,牙齿发育异常,11月龄时曾因肺动脉瓣狭窄行球囊扩张术,术前肺动脉跨瓣压差81 mmHg,术后61 mmHg。超声心动图示:双侧心室壁肥厚,均可见粗大肌小梁,室间隔最厚处14.6 mm,左室后壁厚13.2 mm,左室流出道收缩期内径略窄,最大流速150 cm/s,左室舒张末径19 mm,收缩末径8 mm,射血分数0.88。右室壁厚度9.5 mm,右室流出道收缩期内径轻度狭窄,最大流速137 cm/s,肺动脉瓣重度狭窄,肺动脉瓣上最大流速472 cm/s,压差89 mmHg。卵圆孔未闭4 mm,心包积液中量。基因突变检测结果:患儿3号染色体RAF1基因的第6外显子区域检到一杂合的c.770C>T碱基取代,该突变为新发突变,导致该蛋白第257位的丝氨酸改变为亮氨酸(p.257,S>L)。未检出与肥厚型心肌病相关的其他已知基因突变。
5例患儿基因突变位点均位于已报道的可导致心肌肥厚的基因结构域上,其中3个基因突变位点(12号染色体PTPN11基因,1号染色体RIT1基因及3号染色体RAF1基因)与肥厚型心肌病相关。
Pubmed数据库检索84篇文献报道其与肥厚型心肌病相关,其中有7例基因突变位点位于PTP结构域,氨基酸改变均为第510位谷氨酰胺改变为谷氨酸(p.510,Q>E),与本组例1、3患儿的突变位点位于同一蛋白结构域,推测其可能与文献报道中的位点具有相同的致病机制。文献检索肥厚型心肌病患儿均有左室流出道梗阻表现,预后不良,与本组例1、3表现相近。
Pubmed数据库检索19篇文献报道其与肥厚型心肌病相关,其中有2例基因突变位点位于G2和Switch Ⅰ结构域,与本组例2患儿突变位点所在结构域相同。
Pubmed数据库检索37篇文献报道其与肥厚型心肌病相关,其中有7例基因突变位点位于CR2结构域,4例报道氨基酸改变为第257位的丝氨酸改变为亮氨酸(p.257,S>L),与本组例4、5突变位点完全相同。
目前认为,Noonan综合征发病与丝裂原活化蛋白激酶信号传导通路(RAS-mitogen-activated protein kinase, RAS.MAPK)的信号上调相关[3]。该通路存在于大多数细胞内,将生长因子、细胞因子等细胞外信号转导至细胞内,促进细胞的增殖、分化、代谢等。当细胞膜表面受体与刺激信号分子结合后,激活Src同源性蛋白2(SHP2),经生长因子受体结合蛋白2(GRB2)募集,与鸟苷酸交换因子(SOS1)形成复合体,使GDP-RAS转化为具有活性的GTP-RAS,活化的RAS蛋白通过一系列的磷酸化反应引起RAF-MEK-ERK信号级联反应,最终ERK信号分子进入细胞核内调节相关基因的转录并对刺激信号作出反应。该信号通路被过度激活时,往往会导致出现细胞增殖与分化异常,从而出现一系列的临床综合征。
Tartaglia等[4]对Noonan综合征患者的PTPN11基因进行测序,发现其中错义突变占到50%。后续多个研究进一步证实PTPN11基因为Noonan综合征的一个主要致病基因,占整体报道的30%~60%[5]。PTPN11基因位于12号染色体长臂,是RAS-MAPK级联反应中的重要元素。突变位点主要集中在3号外显子编码的N-SH2区和8、13号外显子编码的PTP区,临床上通常表现为肺动脉狭窄、房间隔缺损、身材矮小、隐睾等,其中有研究表明,携带PTPN11基因突变的NS患者肺动脉狭窄的发生率可达45%~70%[5]。本组例1、3基因突变分别位于PTPN11基因PTP结构域的第13外显子(p.506,Q>P)及第7外显子(p.279,Y>C)。
Razzaque等[6]报道RAF1基因突变在Noonan综合征中可能导致肥厚型心肌病。文章中提出,RAF1是一种RAS信号传导途径下游的效应器,在整个过程中发挥非常重要的调节作用。RAF1基因位于3号染色体长臂,由17个外显子组成,编码区主要有CR1,CR2,CR3三组结构域,CR1包括RAS蛋白结合位点和半胱氨酸富集位点,研究认为上述两处位点通过物理相互作用参与RAF1的负调节;CR2包含了丝氨酸259残端,其对于RAF1的自我抑制功能有着重要的作用;CR3在信号传导途径中负责调控催化剂活性。上述3组结构域通过维持14-3-3蛋白聚体与磷酸化丝氨酸259及丝氨酸621残端的结合,承担了RAF1基因的负调节功能,当结构域产生突变时,将会导致RAF1基因自我抑制功能障碍,进而激活RAS.MAPK级联反应。本组例4、5的基因突变均位于RAF1基因CR2结构域的第7外显子(p.257,S>L)。
Aoki等[7]对180例Noonan综合征患者的基因突变进行了检测,其中17例携带RIT1基因突变,共发现9种错义突变形式,报道了RIT1基因突变可导致Noonan综合征,并提出其突变发生率可能与RAF1基因突变相似。上述结论也在2014年Bertola等[8]的研究中被证实。RIT1位于1号染色体长臂,由6个外显子组成,主要包括G1、G2、Switch Ⅰ和Switch Ⅱ等几个结构域,负责编码RAS亚族属的一种小型GTP酶,与RAS蛋白有50%的同源性,其中Switch Ⅰ和Switch Ⅱ结构域主要负责调解GTP酶的活性。既往研究表明,RIT1基因突变会导致转录因子ELK1的过度激活,从而显著上调了RAS.MAPK信号通路,但各结构域在激活ELK1因子中的作用目前尚未完全阐明。
既往研究表明[9],肥厚型心肌病主要是由编码肌小节或肌小节相关蛋白的基因突变所引起,多为常染色体显性遗传。其中8种参与编码肌小节和心肌驱动分子的基因突变被证实与肥厚型心肌病密切相关,包括MYH7(30%~35%)、MYBPC3(20%~30%)、TNNT2(10%~15%)、TNNI3(<5%)、TPM1(<5%)、ACTC(<1%)、MYL(<1%)[10]。Noonan综合征患者合并心血管系统病变率高达50%~90%,其中合并肥厚型心肌病的比例达20%左右,在已知的Noonan综合征致病基因中,PTPN11基因、SOS1基因、KRAF基因、RAF1基因及RIT1基因均有研究证实其突变可导致肥厚型心肌病,鉴于Noonan综合征与肥厚型心肌病均属于单基因病,推测二者在发病机制上可能存在共同的通路。但关于RAS.MAPK信号通路上调与肌小节编码异常之间的关联目前尚未有文献进行报道。
Noonan综合征的遗传异质性强,不同致病基因合并肥厚型心肌病的比例有很大差异。同一基因突变可有不同临床表现,甚至同一基因同一位点突变临床亦可表现明显差异,本组例4、5相同基因RAF1相同位点突变,但是例5的肺动脉瓣狭窄及心室肌肥厚明显重于例4。在常见的Noonan综合征致病基因中,RAF1被认为与肥厚型心肌病相关性最高,曾有文献报道其突变携带者发生肥厚型心肌病的比例高达80%,且有早发肥厚型心肌病的可能性。本组例1、2、3、4的突变基因虽不同,但均在新生儿期因心脏杂音,超声心动图检查发现心肌肥厚及肺动脉瓣狭窄,例1胎儿心脏超声即怀疑心肌病,最终经基因检测才明确诊断。因此,重视伴肥厚型心肌病的Noonan综合征的基因诊断,对鉴别诊断、早期治疗及遗传生育咨询都有重要的意义。
本组5例均有不同程度的肺动脉瓣狭窄及左、右心室流出道梗阻。但是例1、2、3、5的心肌肥厚程度明显重于例4。前3例患儿同时合并有重度肺动脉瓣狭窄及中度以上左室流出道梗阻,临床症状出现早、进展快、预后不良。PTPN11基因突变的研究中发现,存在p.510,Q>E突变的患者常存在严重的、甚至梗阻型的肥厚型心肌病,与本组例1、3临床特点相符。小鼠实验中证实p.510,Q>E突变导致的SHP2蛋白功能异常,通过影响哺乳动物雷帕霉素靶点(mammalian target of rapamycin,mTOR)信号传导功能导致肥厚型心肌病[11]。随后有报道对p.510,Q>E突变的严重心肌肥厚患儿应用雷帕霉素治疗可改善其心功能及BNP[12],这也为临床治疗带来了新思路。例3心脏外科术后48 h内低心排死亡,如果口服雷帕霉素的基础上行肺动脉瓣球囊扩张,是否更利于患儿的生存,值得临床医生思考。
值得注意的是,本组例1、2的基因突变检测中提示存在有MYPN、MYH6及MYBPC3等肥厚型心肌病相关基因,鉴于上述基因均来源于其无临床表型的母亲,考虑为不完全外显可能性大,但仍不能排除存在多基因协同作用的可能性。目前尚未有文献报道关于PTPN11基因与RIT1基因突变可导致MYPN、MYH6及MYBPC3等杂合基因突变功能异常激活,进而出现特征性临床表型,这部分机制仍需进一步研究关注。
综上所述,Noonan综合征的遗传异质性强,不同突变位点临床表现差异显著。本研究从分子生物学及临床表现分析了PTPN11基因上的p.506,Q>P及p.279,Y>C氨基酸改变、RAF1基因上的p.257,S>L氨基酸改变以及RIT1基因上的p.41,A>T氨基酸改变与Noonan综合征合并肥厚型心肌病的相关性,其中携带PTPN11基因PTP结构域内位点突变往往导致严重的梗阻型心肌肥厚而预后不良。基因检测提高了Noonan综合征的诊断水平,为基因的分子生物学研究临床转化精准治疗提供了可能性。
