巨噬细胞极化在早期急性呼吸窘迫综合征炎症反应中的作用
2019年6月
中华放射学杂志,第58卷第6期 第472页-第475页
张实,谢剑锋,邱海波
急性呼吸窘迫综合征(ARDS)是重症医学科常见的疾病[1],严重ARDS患者病死率为40%~46%。ARDS的危险因素包括肺内因素和肺外因素[2]。肺部促炎/抗炎失衡是早期ARDS发病的枢纽,而巨噬细胞极化在促炎/抗炎中发挥重要作用。现对巨噬细胞极化在早期ARDS肺部炎症中的作用综述如下。
炎症失衡是ARDS发病机制的本质。动物实验表明[3,4,5],脂多糖、油酸等不同的ARDS模型,均造成肺部炎症失衡和炎症性损伤;同时,抗炎相关的介入手段可有效降低动物模型肺损伤程度及死亡数。临床研究亦证实[6],肺内外ARDS患者,肺泡灌洗液中炎性因子水平升高。可见,炎症反应是ARDS发病的主要机制。
然而,抗炎治疗的临床试验遇到瓶颈。糖皮质激素[7]、免疫调节型肠内营养[8]、乌司他丁[9]等抗炎手段均未能改善ARDS患者病死率。有学者认为[1],抗炎治疗失败的原因是患者的选择及抗炎的时机不当。不同病因的ARDS,其炎症机制可能不同。因此,对ARDS炎症机制的深入研究,可能为个体化抗炎治疗提供思路。
炎症反应的机制是炎性细胞发挥生物学功能的过程。炎性细胞产生多种炎性介质和细胞因子,导致大量中性粒细胞在肺内聚集、激活,并通过"呼吸爆发"释放氧自由基、蛋白酶和炎性介质,引起靶细胞损害,表现为肺毛细血管内皮细胞和肺泡上皮细胞损伤,肺微血管通透性增高和微血栓形成,大量富含蛋白质和纤维蛋白的液体渗出至肺间质和肺泡,形成非心源性肺水肿,透明膜形成,以及肺间质纤维化[10]。
参与ARDS早期炎症调控的主要细胞包括,巨噬细胞、中性粒细胞、肺泡Ⅱ型上皮细胞等。肺内巨噬细胞被激活后,释放大量促炎因子和趋化因子,招募其他免疫细胞进入肺内,并与其相互作用,启动肺部炎症。中性粒细胞招募至肺部,释放一些有毒介质,包括蛋白酶、活性氧、促炎细胞因子和促凝血的分子,导致肺上皮-内皮屏障功能损伤。肺泡Ⅱ型上皮细胞被肿瘤坏死因子α(TNFα)激活后,分泌趋化因子,继续招募单核细胞和中性粒细胞,维持肺部炎症反应。由此可见,虽然中性粒细胞和肺泡Ⅱ型上皮细胞均发挥作用,但最初启动肺部炎症的细胞是巨噬细胞。因此,了解巨噬细胞在ARDS发病机制中的作用,是理解ARDS炎症失衡机制的关键。
ARDS初期,肺泡巨噬细胞转化为M1型巨噬细胞。巨噬细胞按其功能分为促炎型-M1型巨噬细胞和抗炎型-M2型巨噬细胞。一些研究证明[11],在小鼠骨髓来源的巨噬细胞培养基中加入内毒素或干扰素γ刺激24 h后,可诱导巨噬细胞极化为M1型。该类细胞为扁圆形,分泌诱导型一氧化氮合酶(iNOS)、TNFα等炎性介质。因此部分学者推断[11],ARDS初期,肺内外损伤因素可能通过病原相关分子模式(PAMP)和危险相关分子模式(DAMP),导致肺内脂多糖和干扰素γ表达上调,诱导肺泡巨噬细胞由静息状态极化为M1型巨噬细胞(促炎型),发挥抗炎作用,其促炎的机制包括分泌促炎因子和趋化因子。
M1型巨噬细胞分泌促炎因子,导致肺部促炎/抗炎失衡。M1型巨噬细胞分泌白细胞介素(IL)-1、TNFα、IL-6、IL-8、IL-12、iNOS等促炎因子[12],这些炎性因子一方面作用于巨噬细胞自身,激活更多的M1型巨噬细胞;另一方面,这些细胞因子与上皮细胞、淋巴细胞和中性粒细胞相互作用,推进肺部炎症反应的进程[13,14]。此外,TNF可上调组织因子,进而促进血小板聚集和微血栓形成,以及肺泡内凝血和透明膜形成。M1型巨噬细胞在肺内分泌的促炎因子,是ARDS初期炎症失衡的关键。
M1型巨噬细胞分泌或上调趋化因子,进一步加重促炎/抗炎失衡。M1型巨噬细胞可分泌趋化因子,如巨噬细胞炎性蛋白2(MIP-2)和角化细胞起源趋化因子(KC)招募中性粒细胞至肺泡。招募的中性粒细胞通过释放有毒介质进一步造成损伤。损伤将破坏肺泡上皮细胞和内皮细胞,导致其屏障功能丧失、通透性增加,进而导致肺泡和肺间质中蓄积富含蛋白的水肿液(肺水肿)。此外,M1型巨噬细胞分泌的TNFα可上调肺泡上皮细胞(AECs)分泌趋化因子继续招募中性粒细胞,维持肺部炎症反应。M1型巨噬细胞起源的趋化因子,持续招募炎性细胞,加重肺部炎症。
ARDS初期,大量巨噬细胞极化为M1型巨噬细胞,分泌大量促炎因子和趋化因子,这是ARDS促炎过重的重要机制。
ARDS初期,肺部内环境缺乏M2型巨噬细胞的诱导剂和增敏剂。研究表明[13,14],小鼠骨髓来源的巨噬细胞培养基中加入IL-4和IL-13可在体外诱导巨噬细胞极化为M2型巨噬细胞。IL-10、转化生长因子β(TGFβ)等一些抗炎因子可增加巨噬细胞表面IL-4受体的密度,使M2型巨噬细胞对IL-4和IL-13的敏感性增加,更容易极化为M2型巨噬细胞。因此部分学者推断[15],ARDS初期,由于M2型巨噬细胞的极化诱导因子(IL-4和IL-13)和增敏因子(IL-10等)不足,导致M2型巨噬细胞数量过少。然而,M2型巨噬细胞是主要的抗炎细胞,可通过三种方式发挥抗炎作用,M2型巨噬细胞的缺乏直接导致肺部抗炎作用不足。
M2型巨噬细胞的胞葬作用可消灭中性粒细胞。Steinberg等[16]证实,脓毒症介导的ARDS患者,肺泡巨噬细胞数与中性粒细胞数呈负相关,与患者不良预后呈负相关。肺内中性粒细胞的清除,主要依靠M2型巨噬细胞。而中性粒细胞浸润,是ARDS炎症损伤的关键问题。因此,M2型巨噬细胞的胞葬作用起到肺部抗炎作用。
M2型巨噬细胞减少炎性细胞招募。M2型巨噬细胞分泌一些基质金属蛋白酶(MMPs)[17],有裂解趋化因子巨噬细胞炎性蛋白2(MIP-2)和趋化因子2(CCL2)的作用,减少中性粒细胞等在肺内的招募。中性粒细胞招募减少,ARDS炎症反应将减轻。因此,M2型巨噬细胞可通过裂解趋化因子,终止炎性细胞招募至肺部,降低肺部炎症反应。
同时,M2型巨噬细胞分泌一些抗炎因子,可中和肺部炎症反应。这些抗炎因子包括白细胞介素受体拮抗剂(IL-1ra)、IL-10、精氨酸酶1(Arg1)等。部分因子通过中和或阻断炎性因子终止其功能(IL-1ra);部分因子通过增加巨噬细胞表面IL-4受体的密度,增强巨噬细胞对IL-4和IL-13的敏感性,利于巨噬细胞极化为M2型巨噬细胞(IL-10)。一些动物实验证明[18],抑制巨噬细胞向M2型极化,将导致持久不停息的炎症反应。因此,M2型巨噬细胞起源的抗炎因子在ARDS中发挥重要抗炎作用。
ARDS初期,肺泡巨噬细胞主要极化为促炎型-M1型巨噬细胞,很少极化为抗炎型-M2型巨噬细胞,这将导致M1/M2比例失衡。这是ARDS促炎/抗炎失衡的重要机制。因此,研究M1/M2极化的调控因素,可能为调控巨噬细胞的极化方向和M1/M2比例提供帮助。
Murray[19]将调控巨噬细胞极化的影响因素分为"完全丧失表型"因子、"逆转表型"因子、"表型调控"因子(
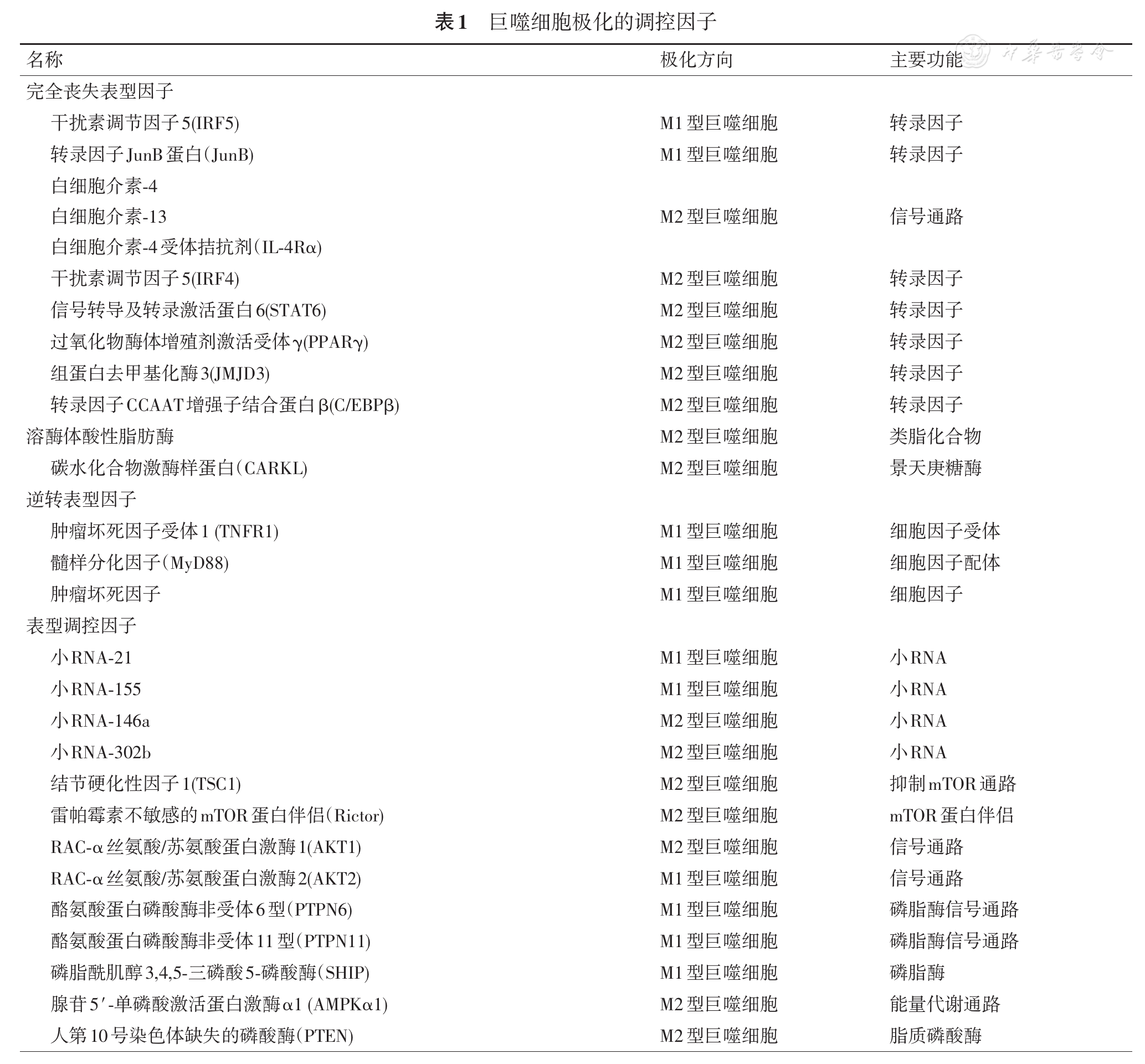
"完全丧失表型"因子是指编码这些因子的基因如果失去功能,会导致M1或M2表型大量缺失。其中M2型巨噬细胞的"完全丧失表型"因子包括IL-4、IL-13、IL-4R、STAT6(转录激活因子6)以及其下游关键的转录因子:干扰素调控因子4(IRF4)、组蛋白去甲基化酶(JMJD3)、过氧化物酶体增殖剂激活受体(PPAR)。Vannella等[20]的研究表明,在IL-4R敲基因小鼠体内,无法诱导M2型巨噬细胞,Arg1等M2型巨噬细胞的生物标志物显著低于野生小鼠。因此推断,IL-4R通路的配体和受体是M2型巨噬细胞极化的必要条件。Satoh等[21]将M2型巨噬细胞诱导剂壳多糖,通过腹腔注入JMJD3敲基因小鼠和野生小鼠,结果发现,JMJD3敲基因小鼠体内几乎不表达M2型巨噬细胞的生物标志物,而野生小鼠M2型巨噬细胞的生物标志物正常表达。M1型巨噬细胞的"完全丧失表型"因子比较模糊,这是因为介导M1型巨噬细胞极化的因素比较多,而哪些因子是调控巨噬细胞由M0型向M1型极化的核心尚不清楚,可能因炎症环境的不同而不同[22]。
"逆转表型"因子是指,随着该因子的消耗,会导致巨噬细胞从M1型逐渐向M2型转换,主要包括TNF及其受体。Kratochvill等[23]的研究表明,小鼠体内TNF表达量与M2型巨噬细胞相关基因表达量呈负相关,与M1型巨噬细胞相关基因表达量呈正相关。并证实,TNF是M2型巨噬细胞极化的重要拮抗剂,其机制是广泛的,既能抑制M2型巨噬细胞激活剂IL-13产生,又能下调M2型巨噬细胞必要转录因子信号转导及转录激活蛋白6(STAT6)的表达。这种调控作用类似于巨噬细胞极化的"开关"。
"表型调控"因子能影响巨噬细胞极化,但无绝对效应。包括小RNA、一些有磷酸化作用的酶和代谢调节因子,主要从转录、蛋白磷酸化、代谢等几个方面调控。Xiao等[24]的研究表明,miR-155与C/EBP基因的转录本结合,抑制C/EBP蛋白表达,导致M1型巨噬细胞增加,促进炎症反应。Zhu等[25]和Wang等[26]的研究表明,let-7e和miR-302b分别通过抑制Toll样受体4及IL-1受体激活蛋白激酶4,介导M2型巨噬细胞增加和抗炎作用。
ARDS早期,调控上述因子可能有助于平衡M1/M2比例,改善促炎/抗炎失衡状态。但"完全丧失表型"因子的作用过于强烈,可能导致新的M1/M2比例失衡;"逆转表型"因子涉及的机制较多,是否有严重的副作用不得而知;"表型调控"因子作用轻微,不同病因的炎症能否起到调控作用仍未知。因此,仍需研究明确不同病因的炎症,上述因子是否能发挥作用,以及治疗时机、可能的副作用等。
ARDS初期,肺内促炎/抗炎失衡,其重要机制是M1/比例失衡。M1型巨噬细胞主要通过分泌促炎因子和趋化因子介导肺部炎症;而M2型巨噬细胞主要通过胞葬作用、裂解趋化因子和分泌抗炎因子发挥抗炎作用。通过影响巨噬细胞极化的因素,保持M1/M2的平衡,可能是ARDS免疫调节治疗新领域。巨噬细胞极化的影响因素分为"完全丧失表型"因子、"逆转表型"因子、"表型调控"因子。调控上述三类因子,可调控M1/M2比例,最终影响机体促炎/抗炎平衡,是ARDS免疫调节治疗有前景的研究方向。
[1] Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment[J]. JAMA, 2018,319(7):698-710.
[2] Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome[J]. N Engl J Med, 2017,377(6):562-572.
[3] Zhang S, Jiang W, Ma L, et al. Nrf2 transfection enhances the efficacy of human amniotic mesenchymal stem cells to repair lung injury induced by lipopolysaccharide[J]. J Cell Biochem, 2018,119(2):1627-1636.
[4] Pan L, Yao DC, Yu YZ, et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats[J]. Biochem Biophys Res Commun, 2016,478(4):1602-1608.
[5] Soncini R, Vieira J, Ramos Lopes AC, et al. Glucocorticoid receptor gene expression in a CLP-induced ARDS-like rat model treated with dexamethasone and metyrapone[J]. Mol Cell Endocrinol, 2018,474:151-157.
[6] Mokra D, Kosutova P. Biomarkers in acute lung injury[J]. Respir Physiol Neurobiol, 2015,209:52-58.
[7] Ruan SY, Lin HH, Huang CT, et al. Exploring the heterogeneity of effects of corticosteroids on acute respiratory distress syndrome: a systematic review and meta-analysis[J]. Crit Care, 2014,18(2):R63.
[8] Chen H, Wang S, Zhao Y, et al. Correlation analysis of omega-3 fatty acids and mortality of sepsis and sepsis-induced ARDS in adults: data from previous randomized controlled trials[J]. Nutr J, 2018,17(1):57.
[9] Leng YX, Yang SG, Song YH, et al. Ulinastatin for acute lung injury and acute respiratory distress syndrome: a systematic review and meta-analysis[J]. World J Crit Care Med, 2014,3(1):34-41.
[10] Urner M, Ferguson ND. A fine balance for oxygen in acute respiratory distress syndrome[J]. Crit Care Med, 2018,46(4):646-647.
[11] Aggarwal NR, King LS, D′Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution[J]. Am J Physiol Lung Cell Mol Physiol, 2014,306(8):L709-725.
[12] Mamedov MR, Scholzen A, Nair RV, et al. A macrophage colony-stimulating-factor-producing γδ T cell subset prevents malarial parasitemic recurrence[J].Immunity,2018,48(2):350-363.e7.
[13] Batra R, Suh MK, Carson JS, et al. IL-1β (interleukin-1β) and TNF-α (tumor necrosis factor-α) impact abdominal aortic aneurysm formation by differential effects on macrophage polarization[J].Arterioscler Thromb Vasc Biol,2018,38(2):457-463.
[14] Luz-Crawford P, Djouad F, Toupet K, et al. Mesenchymal stem cell-derived interleukin 1 receptor antagonist promotes macrophage polarization and inhibits B cell differentiation[J]. Stem Cells, 2016,34(2):483-492.
[15] Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis[J]. Immunity, 2016,44(3):450-462.
[16] Steinberg BE, Goldenberg NM, Laffey JG. Extracorporeal membrane oxygenation for blastomycosis-related severe ARDS: a new indication as a rescue therapy?[J]. Can J Anaesth, 2015,62(7):731-735.
[17] Kaspiris A, Khaldi L, Chronopoulos E, et al. Macrophage-specific metalloelastase (MMP-12) immunoexpression in the osteochondral unit in osteoarthritis correlates with BMI and disease severity[J]. Pathophysiology, 2015,22(3):143-151.
[18] Zhang Z, Xu J, Liu Y, et al. Mouse macrophage specific knockout of SIRT1 influences macrophage polarization and promotes angiotensin II-induced abdominal aortic aneurysm formation[J]. J Genet Genomics, 2018,45(1):25-32.
[19] Murray PJ. Macrophage polarization[J]. Annu Rev Physiol, 2017,79:541-566.
[20] Vannella KM, Barron L, Borthwick LA, et al. Incomplete deletion of IL-4Rα by LysM(Cre) reveals distinct subsets of M2 macrophages controlling inflammation and fibrosis in chronic schistosomiasis[J].PLoS Pathog,2014,10(9):e1004372.
[21] Satoh T, Takeuchi O, Vandenbon A, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection[J]. Nat Immunol, 2010,11(10):936-944.
[22] Lee SG, Oh J, Bong SK, et al. Macrophage polarization and acceleration of atherosclerotic plaques in a swine model[J]. PLoS One, 2018,13(3):e0193005.
[23] Kratochvill F, Neale G, Haverkamp JM, et al. TNF counterbalances the emergence of M2 tumor macrophages[J]. Cell Rep, 2015,12(11):1902-1914.
[24] Xiao Y, Palomero J, Grabowska J, et al. Macrophages and osteoclasts stem from a bipotent progenitor downstream of a macrophage/osteoclast/dendritic cell progenitor[J]. Blood Adv, 2017,1(23):1993-2006.
[25] Zhu W, Yu J, Qiu S, et al. MiR-let-7a regulates anti-citrullinated protein antibody-induced macrophage activation and correlates with the development of experimental rheumatoid arthritis[J]. Int Immunopharmacol, 2017,51:40-46.
[26] Wang Y, Yu T, Jin H, et al. Knockdown MiR-302b alleviates LPS-induced injury by targeting smad3 in C28/I2 chondrocytic cells[J]. Cell Physiol Biochem, 2018,45(2):733-743.
