尾端退化综合征一例
2020年2月
中华放射学杂志,第54卷第2期 第157页-第158页
张毓芬,曾宪春,刘宗才
患儿 男,6个月10 d,因"出生后双下肢畸形并大小便失禁4个月余"就诊。查体:患儿双下肢畸形呈"O"形、双下肢肌肉萎缩、感觉消失伴运动障碍。
DR示骨盆狭小、变形,双侧髂骨内移,骶椎未见显示,双髋关节呈外展位,双膝关节呈屈曲位(
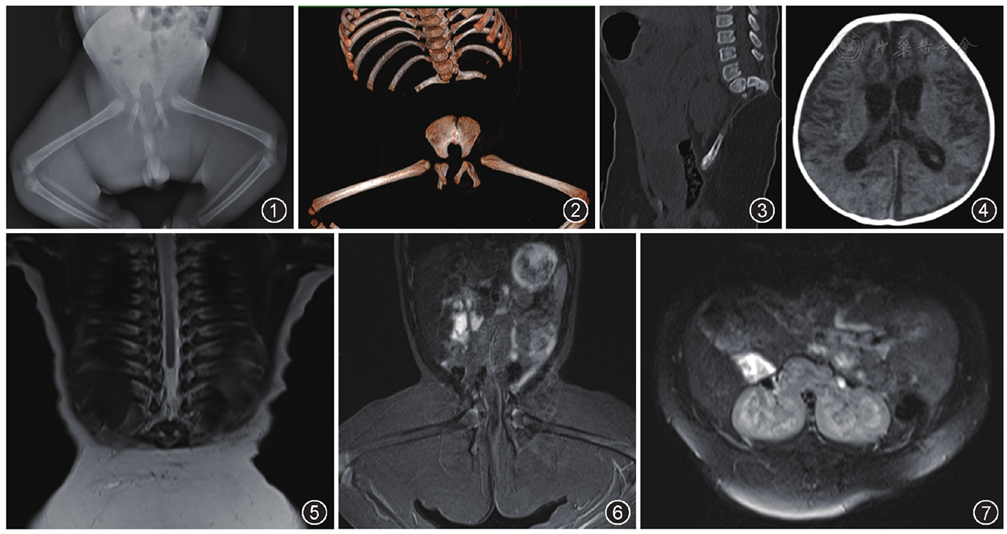
先天性腰骶尾椎缺失是椎管在胚胎时期发育异常或脊神经异常所致,极为罕见,多见于死胎或围产期死亡的新生儿。临床上表现为骨盆狭窄和臀间裂变短,臀部及双下肢肌肉萎缩,随着患儿生长肌肉缺乏更加明显,常合并眼、心血管及脊髓脊膜膨出等畸形。本例患儿合并颅脑发育异常,马蹄肾畸形。骶骨发育不全,又称尾端退化综合征,是一种罕见的先天性畸形,表现为部分或全部骶椎缺如的同时,常合并脊柱脊髓、神经根、泌尿系统、直肠肛门、下肢等多器官畸形,Renshaw分型分为4型[1],本例为第Ⅳ型,完全性骶骨缺如伴不同程度的腰椎发育不良。本例患儿MRI显示脊髓末端位于胸9椎体水平,腰背部、双侧臀部、髋部及双侧下肢肌肉缺失,呈脂肪化。该患儿腰骶尾椎缺如平面较高,由于缺乏脊柱的支撑作用,出现骨盆变形、髂骨内移并形成假关节,并伴随神经肌肉系统异常。根据Guille等[2]对腰骶椎发育不良的分型,本例患儿属于C型。一般本病诊断比较容易,患儿多因腰背部后凸,不能触及腰骶椎,大小便失禁,双下肢畸形就医,依靠X线、CT、MRI等检查可确诊。
[1] Renshaw TS. Sacral agenesis[J]. J Bone Joint Surg Am, 1978, 60(3):373-383.
[2] Guille JT, Benevides R, DeAlba CC, et al. Lumbosacral agenesis: a new classification correlating spinal deformity and ambulatory potential[J]. Bone Surg, 2002,84:32-38.
