[1] Bellaye PS, Kolb M. Why do patients get idiopathic pulmonary fibrosis? Current concepts in the pathogenesis of pulmonary fibrosis[J]. BMC Med, 2015,13:176. DOI: 10.1186/s12916-015-0412-6.
[2] Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management[J]. Am J Respir Crit Care Med, 2011,183(6):788-824. DOI: 10.1164/rccm.2009-040GL.
[3] Wuyts WA, Cavazza A, Rossi G, et al. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic?[J]. Eur Respir Rev, 2014,23(133):308-319. DOI: 10.1183/09059180.00004914.
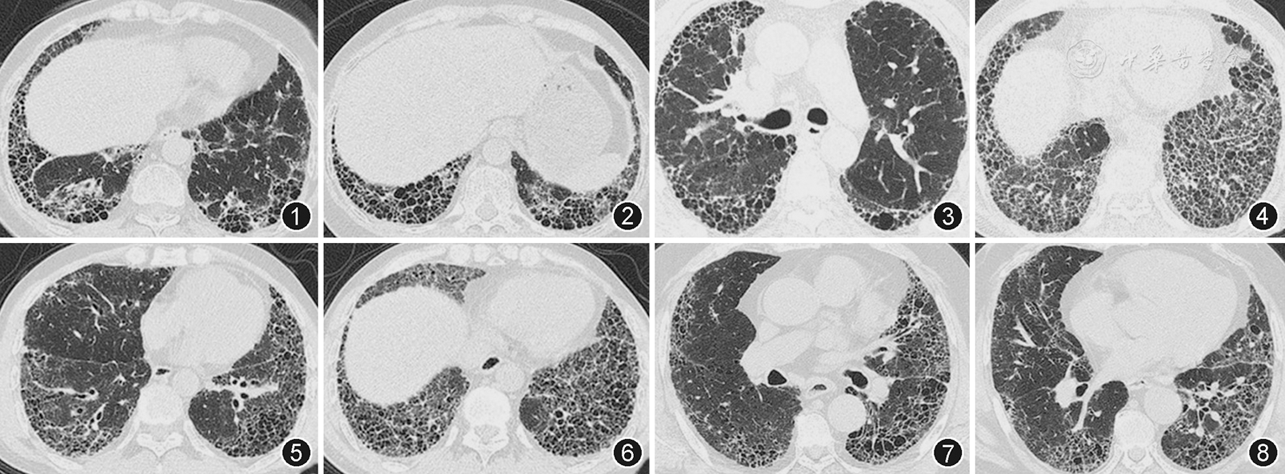
[4] Sumikawa H, Johkoh T, Fujimoto K, et al. Pathologically proved nonspecific interstitial pneumonia: CT pattern analysis as compared with usual interstitial pneumonia CT pattern[J]. Radiology, 2014,272(2):549-556. DOI: 10.1148/radiol.14130853.
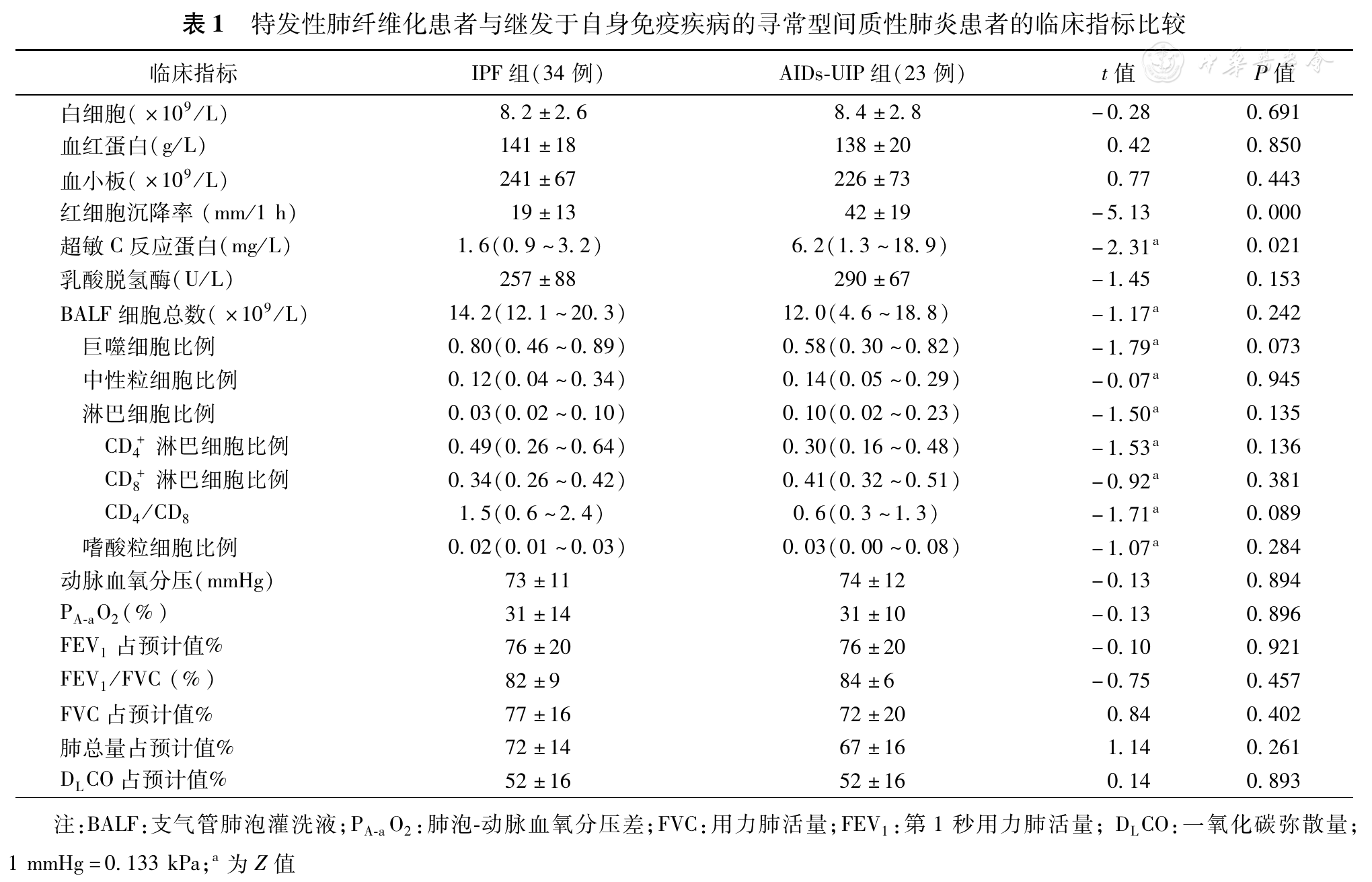
[5] Flaherty KR, Colby TV, Travis WD, et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease[J]. Am J Respir Crit Care Med, 2003,167(10):1410-1415. DOI: 10.1164/rccm.200204-373OC.
[6] Mackay F, Schneider P, Rennert P, et al. BAFF AND APRIL: a tutorial on B cell survival[J]. Annu Rev Immunol, 2003,21:231-264. DOI: 10.1146/annurev.immunol.21.120601.141152.
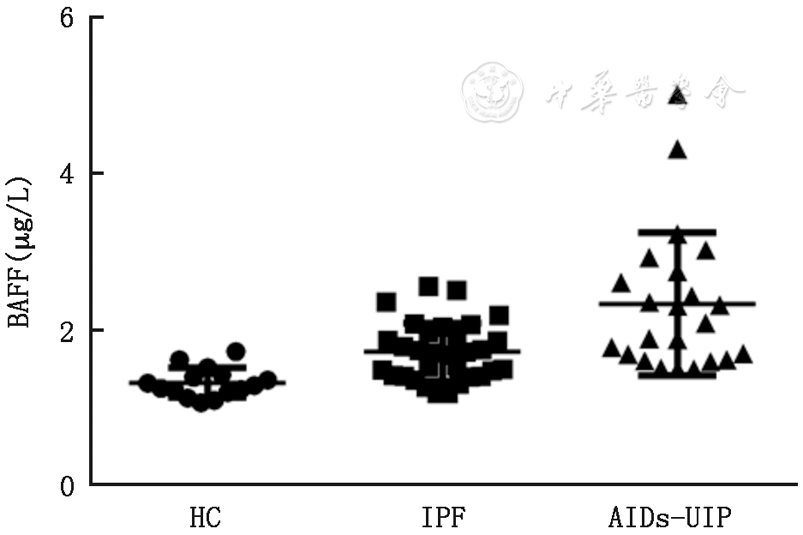
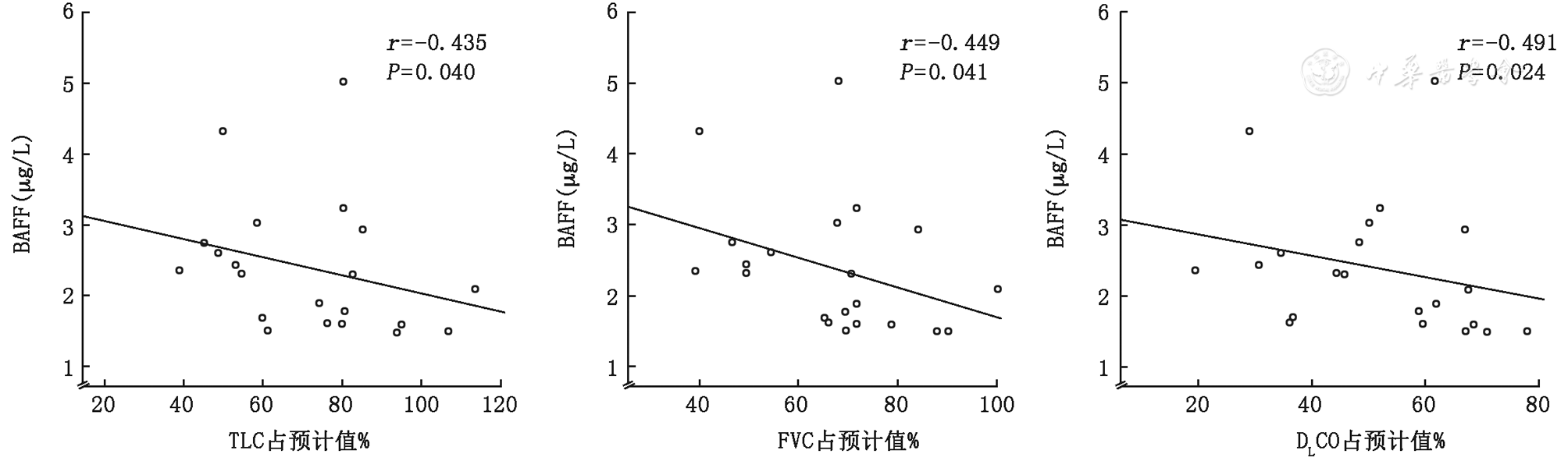
[7] Xue J, Kass DJ, Bon J, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients[J]. J Immunol, 2013,191(5):2089-2095. DOI: 10.4049/jimmunol.1203476.
[8] Drew PA, Takezawa K. Pulmonary cryptococcosis and pituitary Cushing′s disease[J]. Diagn Cytopathol, 1998,18(5):365-367.
[9] Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis[J]. Arthritis Rheum, 1988,31(3):315-324.
[10] Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides[J]. Arthritis Rheum, 2013,65(1):1-11. DOI: 10.1002/art.37715.
[11] Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sj?gren′s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group[J]. Ann Rheum Dis, 2002,61(6):554-558.
[12] Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee[J]. Arthritis Rheum, 1980,23(5):581-590.
[13] Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features[J]. Eur Respir J, 2015,46(4):976-987. DOI: 10.1183/13993003.00150-2015.
[14] Loomis-King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis[J]. Curr Opin Pharmacol, 2013,13(3):377-385. DOI: 10.1016/j.coph.2013.03.015.
[15] Kim EJ, Elicker BM, Maldonado F, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease[J]. Eur Respir J, 2010,35(6):1322-1328. DOI: 10.1183/09031936.00092309.
[16] Wallace B, Vummidi D, Khanna D. Management of connective tissue diseases associated interstitial lung disease: a review of the published literature[J]. Curr Opin Rheumatol, 2016,28(3):236-245. DOI: 10.1097/BOR.0000000000000270.
[17] Romagnoli M, Nannini C, Piciucchi S, et al. Idiopathic nonspecific interstitial pneumonia: an interstitial lung disease associated with autoimmune disorders?[J]. Eur Respir J, 2011,38(2):384-391. DOI: 10.1183/09031936.00094910.
[18] Kono M, Nakamura Y, Enomoto N, et al. Usual interstitial pneumonia preceding collagen vascular disease: a retrospective case control study of patients initially diagnosed with idiopathic pulmonary fibrosis[J]. PLoS One, 2014,9(4):e94775. DOI: 10.1371/journal.pone.0094775.
[19] Hosoda C, Baba T, Hagiwara E, et al. Clinical features of usual interstitial pneumonia with anti-neutrophil cytoplasmic antibody in comparison with idiopathic pulmonary fibrosis[J]. Respirology, 2016,21(5):920-926. DOI: 10.1111/resp.12763.
[20] White ES, Xia M, Murray S, et al. Plasma Surfactant Protein-D, Matrix Metalloproteinase-7, and Osteopontin Index Distinguishes Idiopathic Pulmonary Fibrosis from Other Idiopathic Interstitial Pneumonias[J]. Am J Respir Crit Care Med, 2016,194(10):1242-1251. DOI: 10.1164/rccm.201505-0862OC.
[21] Furukawa H, Oka S, Shimada K, et al. Genetics of Interstitial Lung Disease: Vol de Nuit (Night Flight)[J]. Clin Med Insights Circ Respir Pulm Med, 2015,9(Suppl 1):1-7. DOI: 10.4137/CCRPM.S23283.
[22] Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling[J]. Semin Immunol, 2006,18(5):263-275. DOI: 10.1016/j.smim.2006.04.006.
[23] Cancro MP, D′Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus[J]. J Clin Invest, 2009,119(5):1066-1073. DOI: 10.1172/JCI38010.
[24] Groom J, Kalled SL, Cutler AH, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sj?gren′s syndrome[J]. J Clin Invest, 2002,109(1):59-68. DOI: 10.1172/JCI14121.
[25] Cheema GS, Roschke V, Hilbert DM, et al. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases[J]. Arthritis Rheum, 2001,44(6):1313-1319. DOI: 10.1002/1529-0131(200106)44:6<1313∷AID-ART223>3.0.CO;2-S.
[26] Hoyne GF, Elliott H, Mutsaers SE, et al. Idiopathic pulmonary fibrosis and a role for autoimmunity[J]. Immunol Cell Biol, 2017,95(7):577-583. DOI: 10.1038/icb.2017.22.
[27] Hamada T, Samukawa T, Kumamoto T, et al. Serum B cell-activating factor (BAFF) level in connective tissue disease associated interstitial lung disease[J]. BMC Pulm Med, 2015,15:110. DOI: 10.1186/s12890-015-0105-0.
[28] Donahoe M, Valentine VG, Chien N, et al. Autoantibody-Targeted Treatments for Acute Exacerbations of Idiopathic Pulmonary Fibrosis[J]. PLoS One, 2015,10(6):e0127771. DOI: 10.1371/journal.pone.0127771.
[29] Furie R, Petri M, Zamani O, et al. A phase Ⅲ,randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus[J]. Arthritis Rheum, 2011,63(12):3918-3930. DOI: 10.1002/art.30613.
[30] Lenert A, Lenert P. Current and emerging treatment options for ANCA-associated vasculitis: potential role of belimumab and other BAFF/APRIL targeting agents[J]. Drug Des Devel Ther, 2015,9:333-347. DOI: 10.2147/DDDT.S67264.
[31] Stohl W, Merrill JT, McKay JD, et al. Efficacy and safety of belimumab in patients with rheumatoid arthritis: a phase Ⅱ,randomized, double-blind, placebo-controlled, dose-ranging Study[J]. J Rheumatol, 2013,40(5):579-589. DOI: 10.3899/jrheum.120886.
[32] Fran?ois A, Gombault A, Villeret B, et al. B cell activating factor is central to bleomycin- and IL-17-mediated experimental pulmonary fibrosis[J]. J Autoimmun, 2015,56:1-11. DOI: 10.1016/j.jaut.2014.08.003.