原发性骨质疏松症是以骨量减少、骨组织的显微结构受损,以松质骨骨小梁变细、断裂、数量减少,皮质骨多孔和变薄为特征,以致骨脆性增高、易于发生骨折的一种全身性骨骼疾病。在临床上表现为腰背疼和病理性骨折,主要发生在中老年人,尤其是绝经后妇女。原发性骨质疏松症分为2种亚型,即Ⅰ型和Ⅱ型。Ⅰ型又称绝经后骨质疏松症,Ⅱ型为老年性骨质疏松症。随着人口老年化的进程,骨质疏松发生率呈逐年上升趋势,成为中老年骨痛、骨折及骨折致残的主要原因之一。成骨细胞、破骨细胞和骨细胞之间的协调是维持骨组织稳态的必要条件。骨吸收和骨形成之间不平衡导致一系列的骨疾病如Paget骨病(PDB)、骨质疏松和骨硬化症[1]。最近研究中,参与自噬的蛋白质降解过程在破骨细胞、成骨细胞、骨细胞的功能中有重要作用,说明自噬通路对骨平衡很重要,同时也暗示自噬可能作为一个新的骨疾病病理机制[2]。
自噬由Ashford和Porter在1962年提出,随后科学家们进行一系列研究,日本的Yoshinori Ohsumi团队通过利用酵母进行大量的研究阐明自噬作用的分子机制[3],并获得2016年诺贝尔生理学或医学奖。自噬是一种进化保守的蛋白降解过程,通过泛素蛋白酶系统维持细胞内稳定。受损的细胞器、细胞内病原体及聚合的蛋白被自噬体包裹转运至溶酶体,并与溶酶体融合形成自噬溶酶体被降解。自噬分为3种:巨自噬(macroautophagy)、微自噬(microautophagy)、分子伴侣自噬(chaperone-mediated autophagy),通常所说的自噬是指巨自噬[4]。基础自噬发生在所有的细胞中,其主要作用是清除受损的细胞器及错误折叠蛋白(这个过程也称为质量控制自噬)[5]。质量控制自噬对维持终末分化、长寿细胞如神经元细胞、肌细胞细胞内稳态和功能非常重要,可能对终末分化骨组织细胞特别是长寿骨细胞作用是相似的。在病理状态如营养缺乏、低氧及感染时,自噬能够被诱导增强,形成诱导性自噬,允许被降解的细胞内物质如氨基酸重新利用以延长细胞的存活[2]。自噬参与多种疾病的发生,包括肿瘤、退行性疾病、免疫性疾病、炎症反应等。如伊维菌素是一种广谱抗寄生虫药,能抑制蛋白激酶B-哺乳动物雷帕霉素靶蛋白(protein kinase B-mammalian target of rapamycin, AKT-mTOR)信号通路,引起自噬潮的增加,抑制乳腺癌细胞的生长[6]。甲状腺激素通过诱导死亡相关蛋白激酶2(death-associated protein kinase 2, DAPK2;sequestosome 1)级联反应促进选择性自噬作用,从而抑制二乙基亚酰胺诱导的肝癌变[7]。自噬作用的激活可能是治疗Ullrich型先天性肌营养不良和贝特莱姆肌病(6型胶原蛋白相关的肌病COL6)的新靶点[8]。自噬相关基因ATG(autophagy-related gene,ATG)3依赖的自噬作用使线粒体稳定,对干细胞多能性获得和维持非常重要[9]。在支气管或肺泡上皮细胞中,脂多糖可能通过mTOR激活,下调自噬作用,随后激活NF-κB信号通路,促进急性肺损伤[10]。
自噬过程由一系列ATG编码的蛋白完成。自噬的启动是由UNC-51样激酶1(UNC-51 like autophagy activating kinase 1, ULK1)复合体,包括ULK1、ATG13、ATG101和FIP200形成的复合体裂解mTORC1复合体,从而激活自噬过程。Beclin-1-Vps34复合体,由Beclin-1、Vps34、Vps15和ATG14L组成,参与囊泡成核。自噬体的形成由两个泛素样耦联系统介导:ATG7和ATG10协调作用合成的ATG12-ATG5-ATG16L复合体及LC3-磷脂酰乙醇胺复合体,胞浆中的LC3-Ⅰ脂质化后定位在自噬体膜上,即形成LC3-Ⅱ,后者成为检测自噬体的生物标志。自噬体与溶酶体融合形成自噬溶酶体,降解被吞噬的蛋白质或细胞器,sequestosome-1(也称为p62)参与这个过程[2,3,11,12]。自噬机制详见图1。

成骨细胞是骨形成细胞,起源于多能干细胞,主要功能是合成骨基质(类骨质)及促进随后的矿化作用。矿化需要两个因素——Ⅰ型胶原蛋白和碱性磷酸酶(ALP),这些是成骨细胞活动的标志物。骨形成发生在骨吸收作用之后,与破骨细胞一起参与骨组织重建,可以保证骨组织为适应其功能进行调整,修复微损伤并维持血清钙的稳态[13]。自噬作用对成骨细胞的生存、增殖分化具有重要作用。在人骨髓间充质干细胞向成骨分化过程中,需要增加的线粒体呼吸功能为这个转化提供更多的能量,同时也产生更多的活性氧。而FOXO3诱导自噬作用使细胞内维持正常的活性氧水平[14]。自噬作用对成骨细胞的生存、增殖分化具有重要作用。在成骨细胞体外培养时,发现矿化过程伴随着自噬LC3-Ⅱ显著增加。为了进一步验证自噬与成骨细胞矿化之间的关系,通过小干扰RNA抑制自噬相关蛋白ATG7和BECN1,均使得矿化能力显著下降,矿化结节显著减少。分别取条件敲除成骨细胞ATG5蛋白编码基因的突变鼠与非突变鼠骨片培养,观察到前者骨片矿化能力显著下降,并伴随破骨细胞增加。同时突变鼠的骨容积减少[15]。200 kD的黏着斑激酶家族相互作用蛋白(focal adhesion kinase family interacting protein of 200 kD, FIP200)是哺乳动物自噬ULK1复合体组成成分,其缺陷可导致自噬多个过程受影响,包括P62表达增加,微管相关蛋白1轻链3(microtubule-associated protein 1 light chain 3, LC3)-Ⅱ转化减少,自噬潮受损。成骨细胞FIP200缺陷引起成骨细胞结节形成障碍,矿化能力受抑制,从而抑制成骨细胞的分化,并最终引起骨量减少[16]。在研究具有生物活性的硅纳米粒子对成骨细胞的分化作用与矿化作用时,观察到NP1内化作用激活ERK1/2信号通路引起自噬过程,发现自噬参与成骨细胞分化和矿化作用[17]。AMPK信号通路在成骨细胞自噬中发挥重要作用。在用NO干预的MC3T3-E1成骨样细胞中,自噬通过激活AMPK信号通路激活自噬,减少NO诱导的细胞凋亡[18](如图2A)。在最新的研究发现,成骨细胞的分化需要双相调节AMPK,分化的早期阶段需要激活AMPK,分化中期,抑制AMPK活性是完成分化过程必须的。在分化早期,胰岛素样生长因子(insulin-like growth factor, IGF)-Ⅰ和IGF结合蛋白(IGFBP)-2激活AMPK,激活的AMPK激活自噬作用。在分化中期,激活的AKT下调AMPK活性,mTOR下调ULK-1,抑制自噬作用[19]。该研究与前文研究结果不一致,可能与自噬水平有关。用NO干预成骨细胞,细胞凋亡被诱导,而同时自噬作用显著增强,从而减少细胞凋亡;而在成骨细胞分化过程的生理状态下,调节自噬水平完成细胞分化。自噬相关的多个蛋白参与成骨细胞的分化和矿化,敲除或缺乏都引起成骨细胞的分化功能被抑制。另外,在软骨内成骨中,自噬参与出生后生长点的软骨细胞发展,并且调控Ⅱ型胶原蛋白的分泌。软骨细胞缺乏ATG7的小鼠表现为内质网内Ⅱ型原胶原蛋白的积累,而细胞外基质的纤维网中缺乏ATG7。并发现出生后小鼠软骨细胞自噬的诱导是纤维细胞生长因子(fibroblast growth factor, FGF)18通过与FGF受体(FGFR)4结合,使JNK磷酸化而被激活,并进一步激活自噬起始诱导物VPS34-beclin-1[20]。
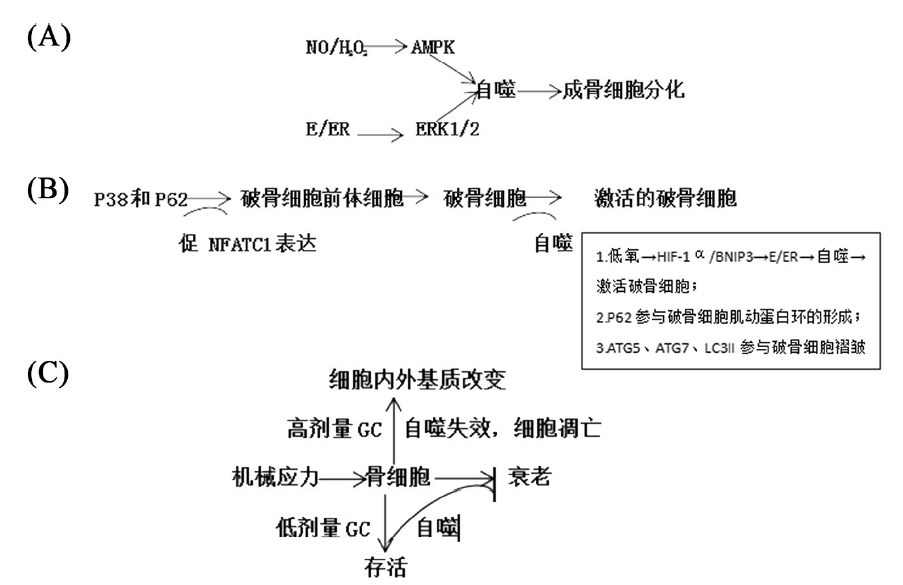

破骨细胞是骨吸收细胞,其衍生于单核/巨噬细胞系的造血细胞。分化的破骨细胞是多核的,其刷状缘贴附在骨组织表面,并分泌溶解和消化骨基质所必须的酸和酶(如基质金属蛋白酶和组织蛋白酶)。酸溶解羟磷灰石,使得蛋白溶解可以接近并降解胶原纤维和其他骨基质蛋白。骨吸收完成后,破骨细胞进入逆转期,发生凋亡,并预示着骨形成的开始[13]。自噬参与破骨细胞形成、分化及骨吸收作用等多个阶段。在破骨细胞前体细胞中,自噬/溶酶体降解肿瘤坏死因子受体相关受体3(tumor necrosis factor receptor-associated receptor 3, TRAF3)是RANKL诱导NF-κB激活的重要一步。自噬/溶酶体抑制剂氯喹通过增加破骨细胞前体细胞TRAF3表达,抑制RANKL诱导的破骨细胞形成[21]。低氧刺激破骨细胞分化,并发现分化的破骨细胞内自噬增加,而抑制自噬作用可以抑制低氧诱导的破骨细胞形成,并进一步发现低氧诱导因子(hypoxia-inducible factor, HIF)-1a及其下游信号分子BNIP3表达增加。将YC-1(HIF-1a特异性抑制剂)与破骨细胞共培养,LC3及破骨细胞均减少,证明HIF-1a/Bcl-2/腺病毒Ela 19 kD结合蛋白3(Bcl-2 adenovirus Ela 19 kD interacting protein 3, BNIP3)信号通路调节自噬活性及低氧条件的破骨细胞分化[22]。ATG5、ATG7、LC3、ATG4B参与破骨细胞极化作用,与破骨细胞皱褶缘形成、溶酶体分泌H+及水解蛋白有关,条件性敲除破骨细胞ATG5、ATG7,导致破骨细胞皱褶缘形成障碍,骨吸收活性减弱[23]。脂化的LC3(即LC3-Ⅱ)依赖ATG5定位在破骨细胞褶皱缘上。ATG5、ATG7和ATG4B以胞浆膜上磷酯酰乙醇胺(phosphatidylethanolamine, PE)耦联的LC为标记,参与溶酶体的靶向运动,并协同作用招募RAS相关GTP结合蛋白7(RAS-related GTP-binding protein 7, Rab7)到褶皱缘发挥作用,进一步调节自噬体和吞噬体的成熟以及吞噬细菌作用。另外推测通过以LC3为特异性靶点,防止溶酶体与细胞内膜的随意结合[24]。P62是高度特异性的自噬适配蛋白,与泛素、LC3结合,调节蛋白聚合物的形成,后被自噬作用降解。在RANKL诱导的破骨细胞形成过程中,发现敲除P62可以抑制破骨细胞的形成。并发现P62参与破骨细胞F-肌动蛋白环(一种破骨细胞分化的标志物)的形成[25]。在条件性敲除p62的骨髓间充质干细胞中,引起NF-κB和p38MAPK、RANKL表达减少,并引起其向破骨细胞分化减少[26]。RANKL触发骨髓巨噬细胞向单核破骨细胞分化,并诱导其向骨表面迁移。达马默德是P38抑制剂,通过抑制P38信号通路,抑制破骨细胞前体细胞NF激活的T细胞c1(NF of activated T cell c1, NFATc1)及迁移与融合相关基因的表达,从而抑制破骨细胞中期分化[27](图2B)。因此推测P62与P38信号通路一起协调RANKL诱导的破骨细胞迁移、融合。
骨细胞由成骨细胞分化而成,并被埋入已矿化的骨基质。骨细胞之间相互连接,与骨表面的成骨细胞也存在连接。连接的渠道是广泛的小管网,小管中含有细胞外液。骨细胞是骨的机械传感器,感受应力和微损伤,并启动相应的骨构建和(或)骨重建[13]。在一项研究中发现纯化的骨细胞表达高水平的RANKL,并且较成骨细胞和骨髓间充质细胞具有更强的促破骨细胞生成作用。另外在骨硬化症表型中观察到缺乏RANKL,特别是骨细胞。因此说明骨细胞是骨重建过程中RANKL的主要来源[28]。在衰老的骨组织中,雷帕霉素通过诱导骨细胞自噬作用,减少年龄相关的骨细胞凋亡,并且通过减少破骨细胞的形成,使骨吸收和骨重建达到平衡[29]。自噬作用可以保护暴露在低剂量的GC的骨细胞,而在高剂量的时候,氧化作用明显增强,细胞失去抗氧化能力,导致骨细胞生存能力降低,引起细胞凋亡。在自噬作用后阶段,自噬作用降解的物质来自自噬溶酶体和自噬泡。这个分解代谢过程释放组织酶和其他水解酶进入细胞浆,在自噬溶酶体与细胞膜融合时,进入周围组织间隙。GC使组织酶和其他水解酶进入细胞基质增加。长时间后引起细胞基质成分改变,并使沉积的矿化基质性质变化。因此,自噬对骨组织基质和骨质量的影响与骨细胞凋亡不同[30]。另外,自噬对机械应力变化高度灵敏。对骨细胞施加生理性应力就可显著诱导自噬,从而减少骨细胞凋亡[31,32](图2C)。然而,机械应力诱导自噬是短暂的,并且是非mTOR依赖的。
随着增龄,骨吸收与骨形成之间平衡被打破,骨强度下降,骨量减少,这些变化最终导致骨质疏松。神经肌肉功能减弱、跌倒是老年相关性骨折的危险因素。衰老引起线粒体功能降低,细胞内活性氧(ROS)增加,导致氧化损伤引起胞内大分子破坏,核、线粒体DNA受损,引起细胞死亡,这是老年性骨质疏松的主要原因[32,33,34]。自噬通过清除受损的线粒体、聚集的蛋白质,减少细胞损害。在氧化应激条件下如在含有H2O2培养基培养,成骨细胞数量减少,自噬增加。H2O2诱导的自噬通过内质网应激(ER)信号通路保护成骨细胞减少其凋亡[35]。在体外培养的MG63成骨细胞中,H2O2诱导AMPK激活、氧化应激、细胞凋亡,激活AMPK信号通路通过维持NADPH,抑制氧化应激,抑制H2O2诱导MG63细胞凋亡,发挥抗凋亡作用,AMPK抑制剂则加重H2O2诱导的成骨细胞死亡[36],说明自噬通过AMPK信号通路抑制H2O2诱导的成骨细胞凋亡。另外一项研究认为衰老的骨组织骨髓脂肪增加,增加脂毒性对骨组织损害。棕榈酸盐(PA)为一种饱和脂肪酸,PA激活自噬和细胞凋亡,引起核裂解。6-氨基-3-甲基嘌呤(6-amino-3-methylpurine, 3MA)是一种自噬抑制剂,抑制PA诱导的自噬体形成,从而减少胞浆细胞色素C表达,抑制成骨细胞凋亡[37]。这项研究显示自噬发挥促细胞凋亡作用,可能与重度自噬损伤细胞有关。雄性SD大鼠随着衰老骨量丢失,骨细胞自噬降低。进一步研究骨细胞自噬与骨丢失之间的关系,发现LC3-Ⅱ阳性细胞与胫骨近端骨密度呈正相关,且与凋亡无明显相关。因此,骨细胞自噬参与年龄相关的骨丢失。在骨质疏松人群中,骨细胞自噬减少,导致年龄相关的骨丢失,且独立于凋亡作用[38]。条件敲除骨细胞ATG7的大鼠,松质骨容积减少、皮质骨变薄、皮质骨筛孔增加、腰椎强度下降,说明抑制骨细胞自噬降低骨量。骨细胞缺乏自噬的大鼠,表现为骨矿化表面减少、骨吸收减少和骨转换减少。抑制骨细胞自噬则增加骨组织氧化应激。这些表现均与衰老引起的骨组织变化相似,所以推测自噬降低可能是衰老对骨量和骨强度的终末效应[39]。总之,自噬在衰老引起的氧化应激中起保护作用,通过增加自噬减轻细胞凋亡。然而,自噬在老年性骨质疏松的确切机制仍有待于进一步研究。
绝经后骨质疏松被认为是绝经后雌激素快速撤退,引起骨吸收作用强于骨形成,导致骨量丢失。雌激素在骨组织中的直接目标是破骨细胞,通过抑制RANKL表达,增加OPG的产生抑制破骨细胞形成。雌激素对成骨细胞作用:抑制氧化应激,成骨细胞凋亡减少,NF-κB活性降低,促进骨形成,另外雌激素参与抑制硬骨素形成[40,41]。特异性敲除雌鼠成骨细胞ERa,在12和18周时,胫骨近端、椎体、股骨远端松质骨骨量减少,胫骨干、股骨远端皮质骨、L5皮质骨骨量减少。胫骨近端松质骨成骨细胞活性减弱,而破骨细胞不受影响。在进行机械检测时,股骨和椎体骨强度降低,说明成骨细胞上ERa对于维持雌性适当骨量和骨强度很重要[42]。而在体外研究中通过血浆剥夺诱导成骨细胞凋亡相关蛋白如caspase蛋白表达上调,细胞凋亡增加、活性减弱,而雌激素通过雌激素受体(ER)增强血浆剥夺诱导的自噬,细胞活性增加,凋亡相关蛋白表达下调,并且LC3-Ⅱ增加。因此认为雌激素通过诱导成骨细胞自噬,抑制其凋亡。进一步观察在血浆剥夺条件下,ERK-1/2、P38、JNK蛋白表达水平及ERK-1/2磷酸化显著增加,因此推测:雌激素通过ER-ERK-mTOR信号轴增强成骨细胞自噬作用,发挥骨保护作用[43]。此外,研究发现HIF-1α/BNIP3信号通路调节自噬活性及低氧条件的破骨细胞分化,而另一项体内研究证明,雌激素缺失能使HIF-1α在破骨细胞稳定存在,参与去势诱导的骨质疏松。在体外敲除ERα的破骨细胞及体内敲除ERα的破骨细胞的小鼠均证明雌激素依赖的骨保护作用需要ERα。另外,通过shRNA同时条件敲除ERα和HIF-1α的WcKO(ERα和HIF-1α敲除)小鼠,其骨密度较ERαcKO(ERα敲除)小鼠增加。骨组织形态学ERαcKO鼠BV/TV、骨小梁数量(Tb.N)减少,骨小梁间隙(Tb.S)增加,而在WcKO都有恢复。说明雌激素/ERα-依赖的HIF-1α降解抑制破骨细胞的自噬作用,抑制破骨细胞形成和骨丢失[44,45]。另外,雌激素/ERα还通过Fas/FasL系统调节破骨细胞凋亡[46]。以上研究都证明雌激素/ERα-自噬信号轴在成骨细胞、破骨细胞中均发挥保护骨组织的重要作用。
去卵巢使大鼠ATG5、LC3和Beclin-1表达增加,P62表达减少,抗氧化标志物总抗氧化能力(T-AOC)、过氧化氢酶(CAT)和过氧化物歧化酶(SOD)活性降低。去势小鼠骨微结构恶化,小梁骨BV/TV、Tb.N减少,Tb.S增大,骨密度显著减少。LC3-Ⅱ阳性细胞率与抗氧化标志物T-AOC、CAT和SOD活性逆相关,BV/TV、Tb.N、骨密度均呈逆相关。E2能使骨量恢复,自噬水平减低,提示去卵巢诱导骨丢失是氧化应激增加、自噬作用增强的结果[47]。一项绝经后妇女的临床研究中,高水平8-羟基-2′-脱氧鸟苷(8-OH-dG)与腰椎、臀骨、股骨颈和转子低骨密度有关。8-OH-dG是调节ROS诱导的嘌呤残基位点及感应DNA氧化损伤的指标,其每增加一个标准差,骨质疏松风险增加54%。而破骨细胞数量及大小都与H2O2呈剂量依赖性增加,过氧化氢酶则抑制破骨细胞形成。人骨髓细胞体外研究显示H2O2可刺激其RANKL、巨噬细胞集落刺激因子(macrophage colony stimulating factor, M-CSF)表达增加。其他研究提示绝经相关的氧化应激与骨密度或骨吸收之间有紧密的联系,绝经后骨质疏松是氧化应激损伤的结果。另外抗氧化应激治疗能够抑制骨丢失相关的疾病[48]。一项绝经晚期骨质疏松临床研究发现与健康对照组相比,绝经晚期骨质疏松患者外周血自噬相关基因上调,成骨细胞形成基因(RUNX2和ALP)下调,并且自噬相关基因的上调与成骨细胞形成基因下调有关,提示绝经晚期骨质疏松骨丢失与骨形成的减少是因为自噬抑制了成骨细胞的形成[49]。这些研究证明自噬增加与骨质疏松有关,这与上文提到自噬参与雌激素保护骨组织结论矛盾,可能是自噬在绝经后不同阶段发挥不同作用,或者自噬在不同的细胞模型发挥作用不同。
总之,自噬在骨质疏松中发挥重要作用是不容置疑的,目前比较明确老年性骨质疏松患者自噬水平低,其机制与细胞凋亡并无明显相关;因此,后续研究可进一步探讨相关的作用机制。然而,绝经后骨质疏松患者自噬水平的变化尚无定论,其作用机制涉及雌激素通过E/ER-ERK-mTOR、HIF-1α/BNIP3-E/ER等信号通路诱导自噬保护骨组织,减少其凋亡。自噬是成骨细胞分化和矿化、破骨细胞激活、骨细胞抗凋亡所必不可少的。自噬在成骨细胞增殖和分化中的作用改变,以及其在形成骨细胞中的作用报道较少,需要进一步研究。加强对自噬的了解可为临床工作进一步了解骨质疏松的发病机制提供思路,也为抗骨质疏松药物的研发提供更多靶点。

原发性骨质疏松症以骨量减少、骨组织的显微结构受损,以松质骨骨小梁变细、断裂、数量减少、皮质骨多孔和变薄为特征。细胞自噬是一种进化保守的蛋白降解过程,通过泛素蛋白酶系统维持细胞内稳定。受损的细胞器、细胞内病原体及聚合的蛋白被自噬体包裹转运至溶酶体,并与溶酶体融合形成自噬溶酶体被降解。自噬参与成骨细胞、破骨细胞形成、分化等多个阶段,在老年性骨质疏松和绝经后骨质疏松中扮演重要角色。