[1] Spath NB, Mills NL, Cruden NL. Novel cardioprotective and regenerative therapies in acute myocardial infarction: a review of recent and ongoing clinical trials[J]. Future Cardiol, 2016,12(6):655-672. DOI: 10.2217/fca-2016-0044.
[2] Hollander MR, de Waard GA, Konijnenberg LS, et al. Dissecting the effects of ischemia and reperfusion on the coronary microcirculation in a rat model of acute myocardial infarction[J]. PLoS One, 2016,11(7):e0157233. DOI: 10.1371/journal.pone.0157233.
[3] Lakota J. Molecular mechanism of ischemia-reperfusion injury after myocardial infarction and its possible targeted treatment[J]. Int J Cardiol, 2016,1(220):571-572. DOI: 10.1016/j.ijcard.2016.06.309.
[4] Rizzo M, Abate N, Chandalia M, et al. Liraglutide reduces oxidative stress and restores heme oxygenase-1 and ghrelin levels in patients with type 2 diabetes: a prospective pilot study[J]. J Clin Endocrinol Metab, 2015,100(2):603-606. DOI: 10.1210/jc.2014-2291.
[5] Okada K, Kotani K, Yagyu H, et al. Effects of treatment with liraglutide on oxidative stress and cardiac natriuretic peptide levels in patients with type 2 diabetes mellitus[J]. Endocrine, 2014,47(3):962-964. DOI: 10.1007/s12020-014-0246-6.
[6] 张军,谷翔,黄问银,等. GLP-1对AGEs诱导H9C2心肌细胞凋亡的保护作用研究[J]. 中国药理学通报,2017,33(1):120-126. DOI: 10.3969/j.issn.1001-1978.2017.01.021.
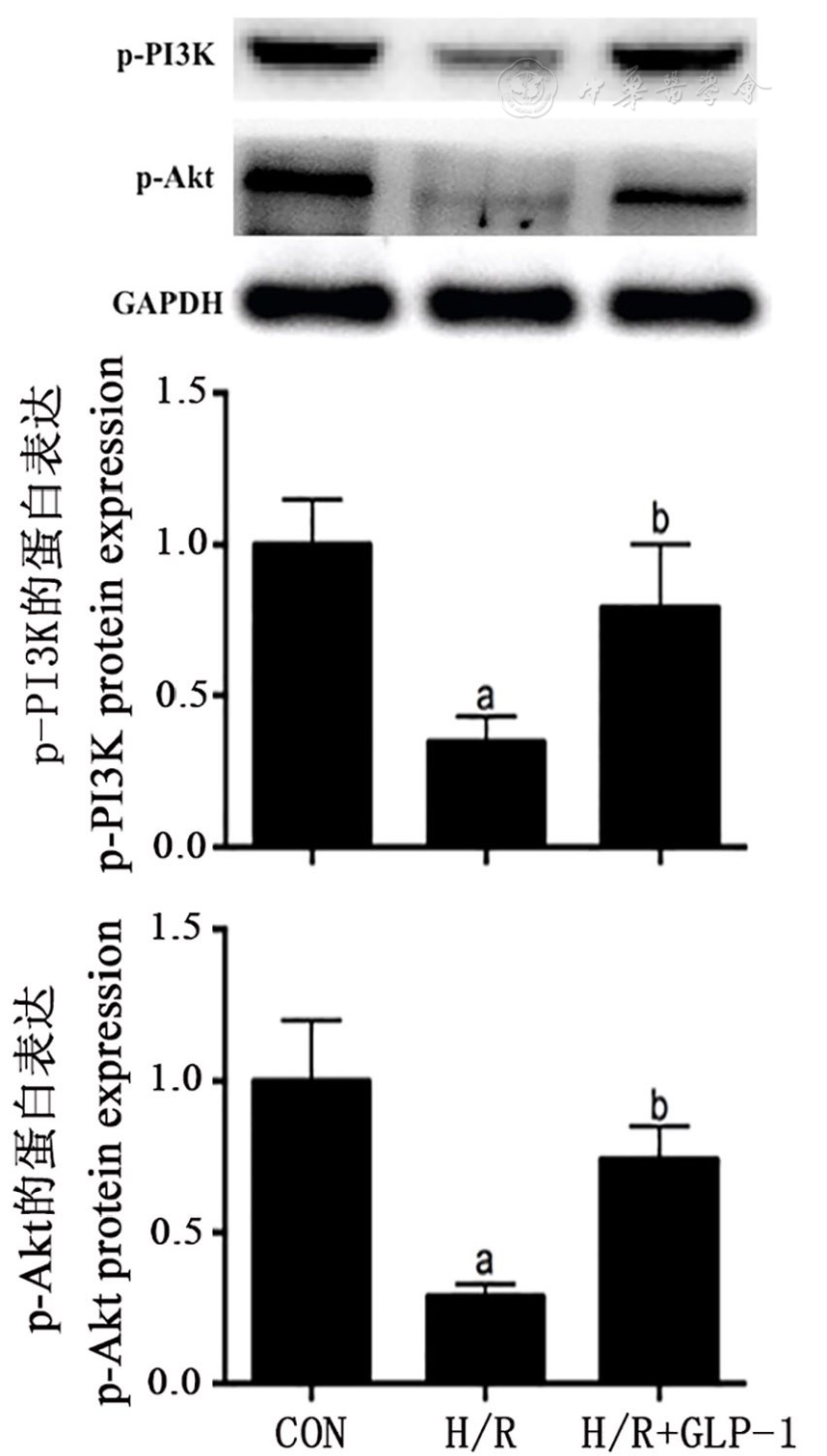
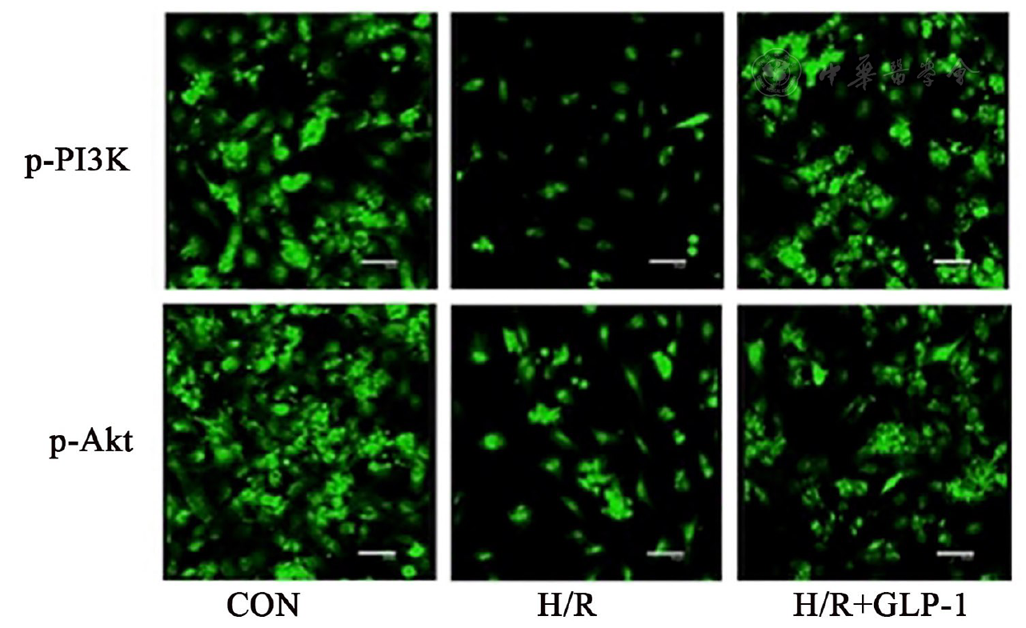
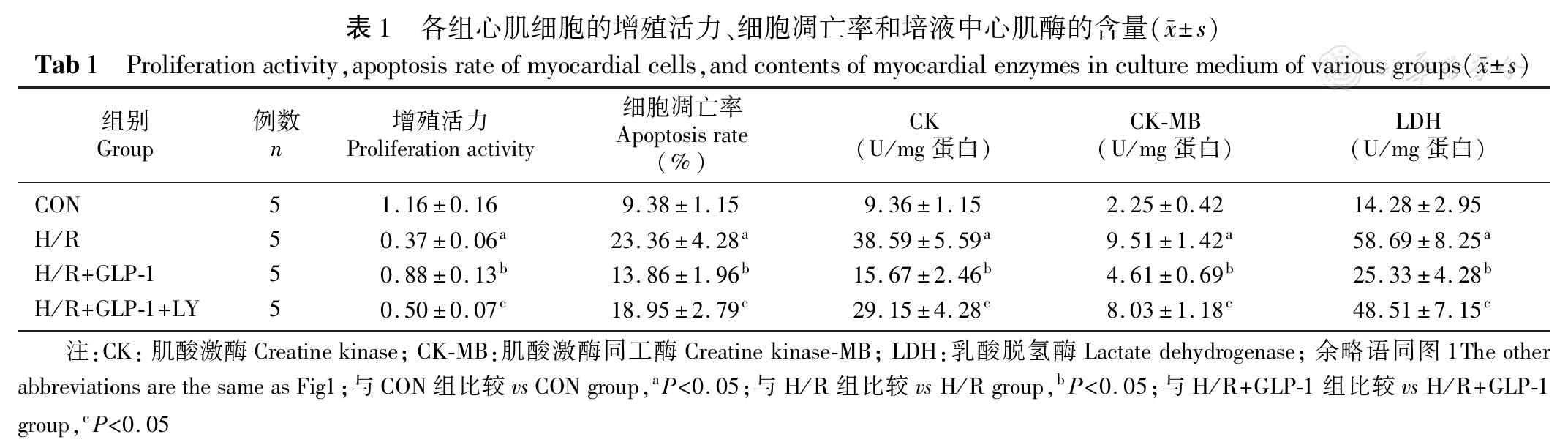
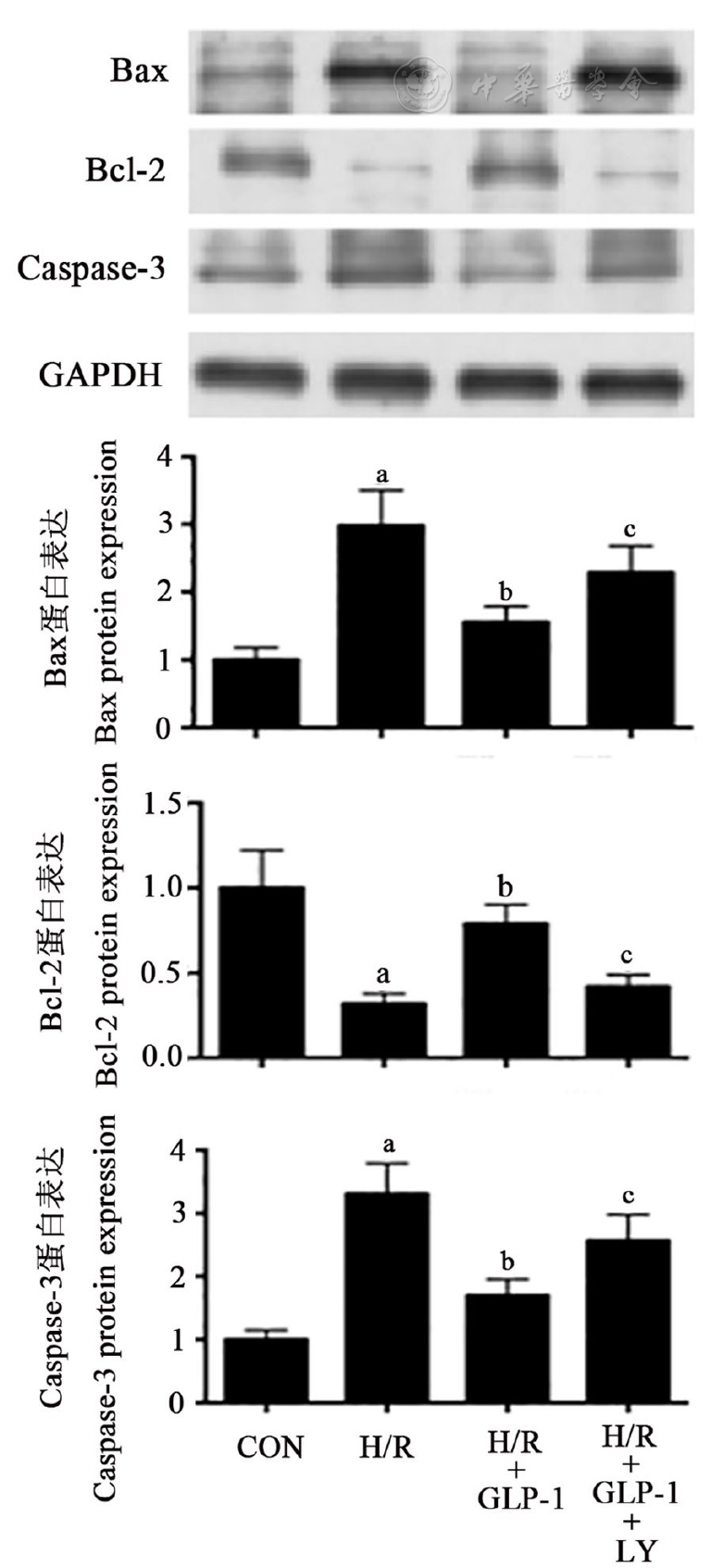
[7] Jiang YQ, Chang GL, Wang Y, et al. Geniposide prevents hypoxia/reoxygenation-induced apoptosis in H9c2 cells: Improvement of mitochondrial dysfunction and activation of GLP-1R and the PI3K/AKT signaling pathway[J]. Cell Physiol Biochem, 2016,39(1):407-421. DOI: 10.1159/000445634.
[8] Engblom H, Heiberg E, Erlinge D, et al. Sample size in clinical cardioprotection trials using myocardial salvage index, infarct size, or biochemical markers as endpoint[J]. J Am Heart Assoc, 2016,5(3):e002708. DOI: 10.1161/JAHA.115.002708.
[9] Bagale KR, Ingle AS, Choudhary R. Contribution of various lipid profile parameters in determining creatine kinase-MB levels in unstable angina patients[J]. Int J Appl Basic Med Res, 2016,6(2):106-110. DOI: 10.4103/2229-516X.179017.
[10] Danese E, Montagnana M. An historical approach to the diagnostic biomarkers of acute coronary syndrome[J]. Ann Transl Med, 2016,4(10):194. DOI: 10.21037/atm.2016.05.19.
[11] Schubert C, Raparelli V, Westphal C, et al. Reduction of apoptosis and preservation of mitochondrial integrity under ischemia/reperfusion injury is mediated by estrogen receptor β[J].Biol Sex Differ, 2016,7:53. DOI: 10.1186/s13293-016-0104-8.
[12] Dong Y, VVR U, Przyklenk K. Inhibition of mitochondrial fission as a molecular target for cardioprotection: critical importance of the timing of treatment[J]. Basic Res Cardiol, 2016,111(5):59. DOI: 10.1007/s00395-016-0578-x.
[13] Su F, Myers VD, Knezevic T, et al. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury[J]. JCI Insight, 2016,1(19):e90931. DOI: 10.1172/jci.insight.90931.
[14] Wang Y, Zhang H, Chai F, et al. The effects of escitalopram on myocardial apoptosis and the expression of Bax and Bcl-2 during myocardial ischemia/reperfusion in a model of rats with depression[J]. BMC Psychiatry, 2014,14:349. DOI: 10.1186/s12888-014-0349-x.
[15] Thomas CJ, Lim NR, Kedikaetswe A, et al. Evidence that the MEK/ERK but not the PI3K/Akt pathway is required for protection from myocardial ischemia-reperfusion injury by 3′,4′-dihydroxyflavonol[J]. Eur J Pharmacol, 2015,758:53-59. DOI: 10.1016/j.ejphar.2015.03.054.
[16] Tobisawa T, Yano T, Tanno M, et al. Insufficient activation of Akt upon reperfusion because of its novel modification by reduced PP2A-B55α contributes to enlargement of infarct size by chronic kidney disease[J]. Basic Res Cardiol, 2017,112(3):31. DOI: 10.1007/s00395-017-0621-6.