中国低血磷性佝偻病/骨软化症诊疗指南
2022年4月
中华内分泌代谢杂志,第38卷第4期 第267页-第281页
徐潮,夏维波,赵家军
低血磷性佝偻病/骨软化症(hypophosphatemic rickets/osteomalacia)是一组由于遗传性或获得性原因导致以低磷血症为主要特征的骨骼矿化障碍性疾病,具有较高的致残、致畸率。发生在儿童期称为佝偻病,主要表现为方颅、鸡胸、肋骨串珠、四肢畸形(下肢膝内翻或膝外翻)、生长迟缓等。成人起病者称为骨软化症,表现为乏力、骨痛,体型改变、身材变矮、多发骨折、活动受限,甚至致残等。
低血磷性佝偻病/骨软化症是常见的代谢性骨病之一,国外报道,发病率为3.9/10万活产新生儿,患病率从1.7/10万儿童到4.8/10万(儿童和成人)不等[1,2,3]。此类疾病在我国的流行病学资料尚待完善。
临床上低磷血症并不少见,其病因包括肠道磷吸收减少、细胞外磷向细胞内转移和肾脏对磷的排泄增加等三个主要方面(


遗传性低血磷性佝偻病/骨软化症包括X连锁遗传(XLH,OMIM#307800、OMIM#300554)、常染色体显性遗传(ADHR,OMIM#193100)、常染色体隐性遗传(ARHR,OMIM#241520、OMIM#613312、OMIM#259775)、伴高钙尿症的遗传性低血磷性佝偻病(HHRH,OMIM#241530)等。其中X连锁低血磷性佝偻病最为常见,在活产婴儿中发病率为1∶20 000,占家族性低血磷性佝偻病的80%以上,由PHEX(phosphate-regulating gene with homology to endopeptidases on the X chromosome)基因异常所致,50%~70%的XLH患者中可检测到PHEX基因突变。
获得性低血磷性佝偻病/骨软化症最常见的原因为肿瘤性骨软化症(tumor induced osteomalacia, TIO)和由于先天因素、其他疾病、药物或毒物引起的肾小管损害或范可尼综合征。
近年来,随着医学遗传学及分子生物学的发展,遗传性低血磷性佝偻病/骨软化症的致病基因陆续被发现,低血磷性佝偻病的发病机制也逐渐被阐明,但是其详细的发病机制还有待于深入研究。作为体内重要的调磷因子:成纤维细胞生长因子23(fibroblast growth factor 23,FGF23)被发现以来,低血磷性佝偻病/骨软化症根据其发病是否由FGF23过多所致,分为FGF23介导的低血磷性佝偻病/骨软化症和非FGF23介导的低血磷性佝偻病/骨软化症两类,分述如下。
FGF23是重要的磷调节因子。主要由成骨细胞及骨细胞分泌,在体内磷稳态维持及维生素D调节方面发挥十分重要的作用。其活性形式为全段FGF23(intact FGF23,iFGF23),当iFGF23被降解为氨基端(nFGF23)和羧基端片段(cFGF23)后则失去生物活性。iFGF-23主要经由肾脏发挥作用,通过抑制肾近端小管钠-磷共转运蛋白2a和2c(NaPi-2a和NaPi-2c)表达,直接抑制肾脏磷的重吸收,导致经肾脏磷丢失增多;另一方面,iFGF-23可以通过抑制1α-羟化酶同时促进24-羟化酶作用,抑制1,25-二羟维生素D[1,25(OH)2D]的生成,进而抑制肠道对磷的吸收[4,5]。
XLH的致病基因是PHEX基因,该基因的失活突变可导致XLH的发生[6,7,8]。PHEX蛋白是M13金属蛋白酶家族成员,其编码基因包含22个外显子,编码蛋白含749个氨基酸。正常PHEX基因编码的PHEX蛋白是一种单跨膜蛋白,属于膜结合的金属蛋白酶家族,与内肽酶家族具有高度的同源性,可降解FGF-23使其失活。XLH患者由于PHEX基因突变,其内肽酶样活性降低,血清中FGF-23的水解灭活受限或生成增加,使多数XLH患者血FGF-23水平升高[9],进而导致肾脏磷排泄增加,1,25(OH)2D的生成减少,导致低磷血症、佝偻病或(和)骨软化症。
由FGF-23基因位于176至179位的RXXR基序上编码精氨酸(R)密码子的杂合突变导致[10]。正常情况下FGF-23分子中Arg179与Ser180分离使FGF-23失活,此区域突变导致FGF-23降解受抑制,血FGF-23水平升高,进而导致低血磷性佝偻病/骨软化症。
ARHR1由编码牙齿和骨骼非胶原基质蛋白的基因(dent matrix protein 1, DMP1)突变所致,该基因突变导致其编码的牙基质蛋白1(DMP1)功能缺失[11]。DMP-1功能缺失可刺激骨细胞分泌FGF-23增加,但作用机制尚不明确。ARHR2是由编码外生核苷酸焦磷酸酶/磷酸二酯酶1(ectonucleotide pyrophosphatase/phosphodiesterase 1,ENPP1)基因失活突变引起,导致其编码的外生核苷酸焦磷酸酶/磷酸二酯酶失活[12]。ENPP1基因突变患者血FGF-23水平升高,但其机制目前尚不明确;ARHR3其致病基因为序列相似20家族成员C(family with sequence similarity 20, member C,FAM20C)基因[13]。已知DMP1的磷酸化有赖于FAM20C,当FAM20C功能缺失时,DMP1部分磷酸化障碍,进而影响FGF-23的代谢。
多数由磷酸盐尿性间叶组织肿瘤分泌过多的FGF23,导致低磷血症和活性维生素D生成减少,进而导致低磷血症和骨软化症。尽管TIO导致FGF23分泌过多的确切机制仍不明确,但近年来仍有较多相关发现和进展,如融合基因(fibronectin-FGF receptor 1、fibronectin-FGF 1)与Klotho/FGF receptor 1复合体在其中的作用等[14]。
通常是由于肾小管功能异常,对磷的重吸收减少,导致低磷血症引起的佝偻病/骨软化症。不同于FGF23介导的低血磷性佝偻病/骨软化症,此类患者的血FGF23水平没有升高,甚至降低。其中HHRH由编码NaPi-2c的SLC34A3基因突变引起。肾小管对磷的重吸收减少,使尿磷酸盐排出增加,患者血清1,25(OH)2D浓度正常或者出现与低磷血症程度相当的升高[15]。由于血1,25(OH)2D水平偏高,肠道钙吸收继发性增加,很可能发生高钙尿症,甚至发生肾结石。其他先天性或获得性的肾小管损害,或范可尼综合征均使肾小管对磷重吸收能力降低,导致低血磷性佝偻病/骨软化症。
低血磷性佝偻病/骨软化症临床异质性强,但当患者具有典型的佝偻病/骨软化症的表现,同时存在低磷血症和明确的家族史时,即可作出初步的临床诊断。进一步进行分子诊断,对制定精准治疗方案、判断疾病预后、临床随诊及产前诊断均至关重要。
低血磷性佝偻病/骨软化症在儿童期和成年后临床表现不尽相同,病情严重程度有异,由于疾病的外显率不同,导致甚至在同一家系成员之间的表型差异[16,17]。
儿童期主要表现为佝偻病,典型的三联征:(1)低磷血症;(2)下肢畸形;(3)生长缓慢。该疾病临床表现轻重不一,轻者仅有低磷血症而无任何骨骼异常。患者常于幼儿起病,1岁前出现枕秃,鸡胸、肋串珠。学步迟缓,步态不稳,四肢短小畸形,下肢畸形,膝内翻、膝外翻或髋内翻。长骨干骺端膨大,出现手足镯征。生长缓慢、身高低于正常同龄儿童。部分患儿伴有囟门关闭延迟及牙齿发育异常,主要表现为出牙延迟甚至牙齿缺失,非龋牙脓肿、釉质缺损、牙髓腔扩大和长冠牙。极少数患儿表现为听力减退、小脑扁桃体下疝等。
成年期主要为骨软化症的表现,包括身材矮、下肢畸形、骨骼痛、假性骨折、关节退行性变及关节炎、肌腱韧带钙化(附着点病),椎管狭窄是一种罕见且严重的晚期并发症,在部分病例中与脊柱纵韧带骨化有关,可致剧痛并严重影响日常活动能力。部分患者反复牙周脓肿和牙齿脱落、听力减退[18,19,20,21,22,23]。
XLH、ADHR和ARHR的临床表现相似,其中ADHR部分患者起病隐匿,有自发缓解的趋势。ADHR临床表型的出现与患者的血清铁水平相关,因此该类疾病在女性中发病率较高,常出现在女性月经初潮、孕产期或围绝经期。ARHR的主要临床表现与XLH相似,但在婴儿时期很难发现,常于幼儿期或成年以后发病,并可能伴有颅骨硬化、广泛骨硬化或全身动脉硬化特殊表现。
肿瘤性骨软化症绝大多数成人起病,主要表现为较严重的四肢无力、行走困难、骨痛、身材变矮、驼背畸形等,容易合并骨折或牙齿脱落。部分患者表现为范可尼综合征样表现,但几乎不出现肾小管酸中毒。
其他几种类型综合征性的低血磷性佝偻病/骨软化症,如McCune-Albright综合征、线样表皮痣综合征(linear nevus sebaceous syndrome)、面骨发育不良(osteoglophonic dysplasia,OGD)和低血磷性佝偻病合并甲状旁腺功能亢进症(hypophosphatemic rickets and hyperparathyroidism)均极为少见,但均会表现为特殊的皮肤或骨骼异常,在临床上容易辨别。
血磷水平显著降低,血钙正常或偏低,尿磷增加,肾磷阈降低,血碱性磷酸酶(alkaline phosphatase, ALP)水平升高,PTH可正常或轻度升高,1,25(OH)2D常较低(见于FGF23相关性低血磷性佝偻病患者),25羟维生素D(25OHD)可正常或偏低,血浆FGF23水平升高[24,25]。
值得注意的是,出生后3~4个月内,血清磷酸盐水平可能在正常范围内[26,27]。应通过计算肾小管磷酸盐最大重吸收量与肾小球滤过率之比(TmP/GFR)评估是否存在肾性失磷[28]。对于磷酸盐摄入量不足或肠道吸收功能受损(可能是由尿磷酸盐水平低造成的)患者,在其血清磷酸盐水平恢复正常之前,TmP/GFR可能存在假性降低。
尽管血浆FGF23水平普遍升高,但在合并低磷血症时,FGF23水平正常并不能排除低血磷性佝偻病,而应解释为不适当之正常[29]。另外,其他因素亦可影响FGF23水平,尤其是应用磷酸盐和维生素D治疗后[2,29,30,31]。因此,FGF23水平对于未经治疗的患者最有价值[32]。
TIO患者可伴有尿氨基酸、尿蛋白阳性或尿糖阳性等范可尼综合征的表现。由于ADHR患者病情活动程度可与铁缺乏相关,建议完善血常规、外周血涂片、血清铁、铁蛋白等指标检查。HHRH患者尿钙水平显著增加。
佝偻病性病变的特征是在长骨干骺端,骨骺的生长板增厚膨出,干骺端增宽似杯状。骨骺端骨小梁紊乱、稀疏粗糙,边缘不齐,呈毛刷样(
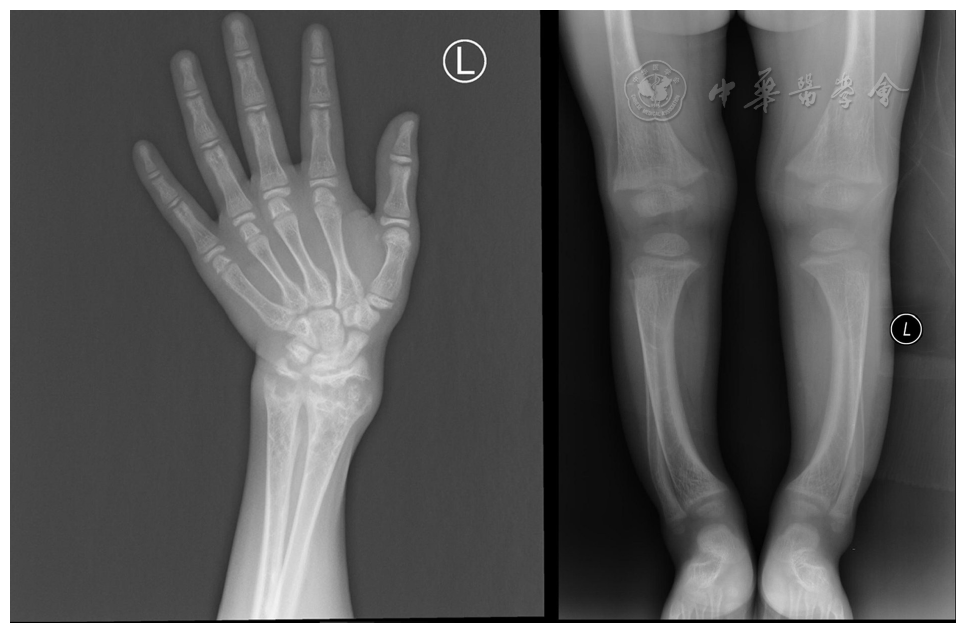
注:X线表现有干骺端增宽,生长板增厚,长骨骺端模糊,呈毛刷征
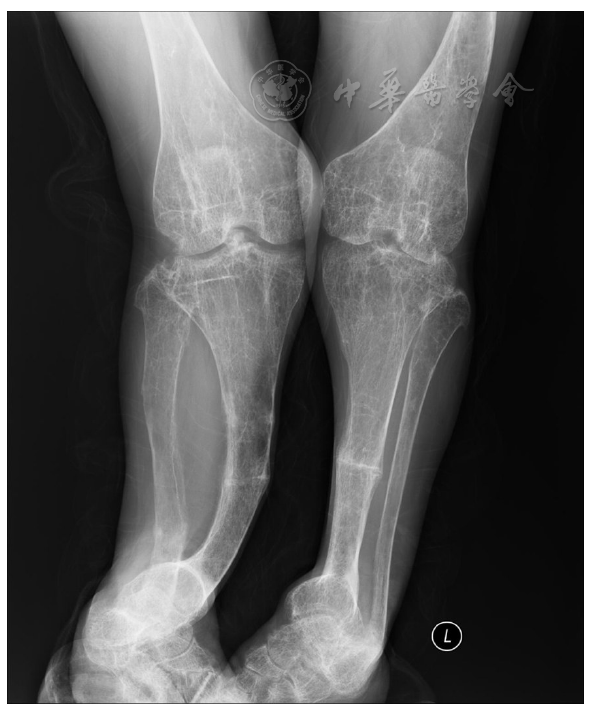
注:X线表现为严重的下肢畸形,长骨的干骺端膨大,胫骨和腓骨严重扭曲
成人可能表现出不同于儿童的影像学特征,骨质稀疏模糊,呈磨玻璃状。容易出现骨骼畸形,有膝内翻或膝外翻,髋臼内陷骨盆呈三叶状,椎体上下缘呈双凹变形。成人XLH患者常表现为脊椎、髋和膝关节早发骨性关节炎(关节边缘骨赘或关节软骨变薄)和(或)附着点病(例如:韧带附着点部位骨增生或韧带钙化)[18,19]。骨软化症相关骨折在成年人中较为罕见,但假性骨折在成年人中却时常可见[16,18,19,33]。最具特征性的是假骨折,一种条状透明区称为Looser区,一般呈对称性分布。
HHRH患者超声检查可能发现泌尿系结石。
对于肿瘤性骨软化症患者,明确定性诊断后应进一步完善生长抑素受体显像、68Ga-DOTATATE-PET/CT检查以寻找肿瘤病灶,对于有阳性发现的局部病灶,应进一步行CT、MRI等影像检查以明确定位诊断。
有典型的X连锁显性遗传家族史应首先建议进行PHEX基因的检测,其他起病较早,或未发现明确的TIO证据者,推荐进行PHEX、FGF23、DMP1、ENPP1、SLC34A3等基因筛查,明确是否存在以上致病基因所致的遗传性低血磷性佝偻病。上述基因筛查阴性时,可进一步行高通量测序(Next-Generation Sequencing,NGS),可为70%~90%的病例提供确切的阴性或阳性结果[34,35,36,37,38,39,40,41,42,43,44,45]。XLH患者部分存在PHEX基因的大片段缺失或插入突变,建议采用多重连接探针扩增(multiplex ligation-dependent probe amplification, MLPA)检测[46,47]。
低血磷性佝偻病/骨软化症的诊断一般可以通过典型的佝偻病/骨软化症的临床表现、低磷血症或TmP/GFR降低、血碱性磷酸酶水平升高和典型的骨骼佝偻病或骨软化症的影像学特征作出诊断。乏力、步态摇摆、上台阶困难等低磷血症的表现往往是低血磷性佝偻病/骨软化症的重要临床线索。
诊断低血磷性佝偻病/骨软化症时需除外由于营养缺乏(维生素D缺乏)或维生素D代谢异常所导致的佝偻病/骨软化症,此类患者通常会以低钙血症为主要特征,血磷仅为轻度降低。需要注意部分低血磷性佝偻病/骨软化症患者可能同时存在维生素D缺乏。
一旦确诊低血磷性佝偻病/骨软化症,下一步应进行低磷血症病因的鉴别。低磷血症同时合并有TmP/GFR降低,提示存在肾小管磷重吸收障碍,多数的遗传性低血磷性佝偻病/骨软化症属于此种类型,包括XLH、ADHR、ARHR、HHRH、TIO和其他综合征性低血磷性佝偻病/骨软化症。XLH、ADHR、ARHR和TIO的临床表现相似,可根据病史和家族史等予以鉴别。其中XLH最为常见,大约占比80%~90%,约半数患者具有典型的X连锁显性遗传的家族史,另半数可能系新发突变;ADHR病情相对较轻、隐匿起病、有自发缓解的趋势、发病多数与缺铁有关,常见于女性月经初潮、孕育期或围绝经期。TIO多见于成人,不具有明确的家族史,低磷血症和骨软化症的病情相对较重。HHRH会有特征性的高尿钙或肾脏及泌尿系结石,有助于作出鉴别诊断。
肠道磷吸收减少和磷向细胞内转移过多所导致的低磷血症,多数通过伴随疾病病史可以作出临床判断,多数为短期的或一过性的,也较少导致佝偻病/骨软化症。
自幼起病的成人低血磷性佝偻病/骨软化症患者会表现为脊柱和关节活动僵直、骨骼X线片提示骨质增生、附着点病、脊柱呈竹节样改变和椎管狭窄等,临床上可能被误诊为强直性脊柱炎、骨性关节炎和脊柱退行性疾病等,应注意鉴别诊断。
低血磷性佝偻病/骨软化症的诊断和鉴别诊断流程见
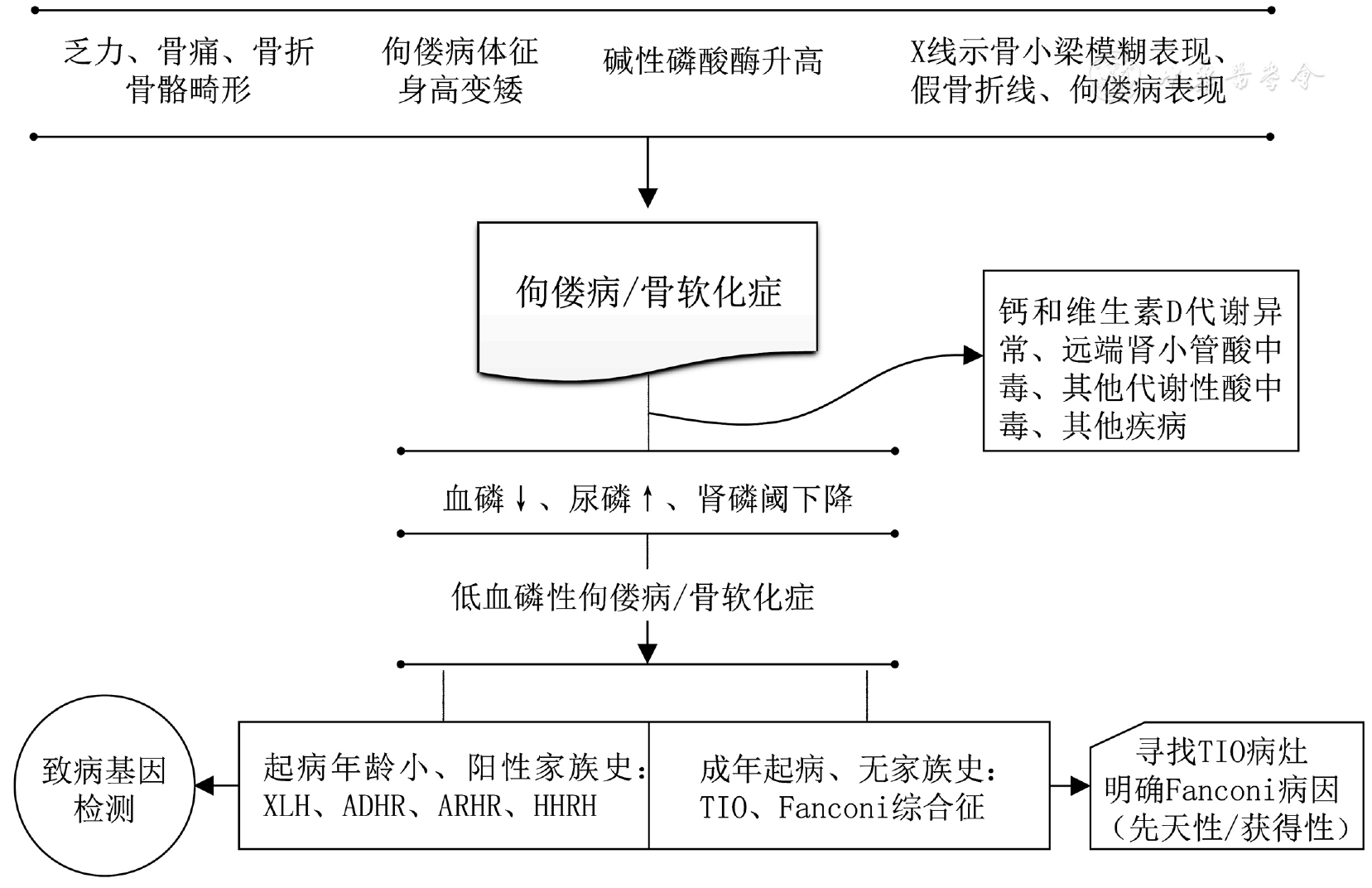
低血磷性佝偻病/骨软化症的治疗目标是纠正低磷血症,改善症状,减少或纠正骨骼畸形,提高生活质量和避免远期并发症和合并症。
针对获得性低血磷性佝偻病/骨软化症,首先需积极确定和去除病因。疑为TIO的患者,需积极寻找导致TIO的责任肿瘤病灶,一旦定位明确,首选手术治疗,手术不能缓解或无法手术的患者可口服磷酸盐(中性磷合剂)和活性维生素D治疗,或可考虑针对FGF23的靶向治疗,如FGF23的单克隆抗体。阿德福韦酯或其他药物及毒物所致范可尼综合征的患者,需停止相关药物或毒物接触。必要时可予以磷酸盐(中性磷合剂)和活性维生素D治疗,如果合并肾小管酸中毒者可同时予以枸橼酸合剂或碳酸氢钠纠正酸中毒。
(1)儿童患者:①磷酸盐(中性磷合剂):磷酸盐补充是低血磷性佝偻病的基本治疗,磷酸盐的治疗剂量因年龄和表型严重程度的不同而不同,目前尚未就口服磷酸盐的最佳剂量达成共识[24,25]。但是,根据表型严重程度,推荐的口服磷酸盐起始剂量(以元素磷计算)为每天20~60 mg/kg(每天0.7~2.0 mmol/kg)。早期治疗者预后更佳[21,23,48,49,50,51,52,53,54,55]。磷酸盐补充剂可以是含有钠盐和(或)钾盐的口服液、胶囊或片剂。由于现有磷酸盐制剂的磷含量差异巨大,因此给药剂量应始终以元素磷含量为基础。我国尚无磷酸盐的胶囊和片剂,多数采用中性磷酸盐缓冲液(或称磷酸盐合剂),常用的配方有:A.磷酸氢二钠(Na2HPO4·12H2O 73 g或Na2HPO4 29 g)+磷酸二氢钾(KH2PO4 6.4 g)加水至1 000 mL,每100 mL中含元素磷779 mg; B. 19.3%磷酸氢二钠和2.05%磷酸二氢钾的混合溶液,每100 mL中含元素磷4.74 g。口服磷制剂后,血磷水平迅速升高,常在60~90 min达到高峰,在4 h内恢复至基线浓度[56]。因此,应尽可能频繁地服用磷酸盐,如每日4~6次,以维持血磷水平较为稳定。口服磷酸盐制剂并不能使空腹磷酸盐水平达到正常,如需监测可考虑在服用磷酸盐后1~1.5 h留取血样测定,但通常不将血磷水平恢复正常作为治疗的目标[24,25]。对于发生骨骼变化前即被确诊的婴儿,其治疗目标是预防佝偻病。低血磷性佝偻病的患者需监测佝偻病的活动程度,目标是使ALP水平降至正常和佝偻病的影像学评分恢复至正常[25,53,57]。同时,患者的骨痛和无力症状减轻,生长加速,下肢畸形纠正和牙齿健康状况改善[21,22,55,56,58,59,60,61,62,63,64,65]。磷酸盐和钙可在肠道内结合沉淀,进而减少前者的吸收,故不应与钙补充剂或钙含量高的食物(如牛奶)同服。由于服用磷酸盐后,血磷一过性升高会刺激PTH的分泌,长期用药可能引起继发性甲状旁腺功能亢进,加重肾性失磷[22,53,64],故口服磷酸盐制剂时,常同时使用活性维生素D。②活性维生素D及其类似物:除口服磷酸盐外,还需服用活性维生素D或其类似物(骨化三醇或阿法骨化醇),目的是纠正体内活性维生素D生成不足,进而预防继发性甲状旁腺功能亢进,增加肠道对磷的吸收。骨化三醇推荐剂量为20~60 ng·kg-1·d-1,分为每天2次用药。阿法骨化醇的等效剂量为骨化三醇的1.5~2.0倍,由于其半衰期较长,可每天1次用药。幼儿期和青春期(生长阶段)所需的剂量较高,可根据血清ALP和PTH水平以及尿钙排泄量来调整剂量。活性维生素D及其类似物可促进生长和骨骼愈合,但会增加高钙尿症和肾钙质沉着的风险[66,67,68,69,70]。反之,活性维生素D剂量不足通常会导致肠道钙吸收降低,尿钙排泄量降低,持续存在的佝偻病以及ALP和(或)PTH水平升高。HHRH的患者不推荐使用活性维生素D及其类似物。③普通维生素D:在活性维生素D应用于临床之前,低血磷性佝偻病/骨软化症的治疗,需要很大量的普通维生素D,每天的用量甚至达到10万~20万IU,因此此类疾病一度被称为"低磷抗维生素D性佝偻病"。但是活性维生素D使用以来,就没必要再使用超大剂量的维生素D来治疗此病。针对部分低血磷性佝偻病/骨软化症可能存在维生素D缺乏,建议使用普通维生素D2或D3制剂补充,使血25羟维生素D的水平保持在30 ng/mL以上。④钙剂:保持患儿的钙摄入量能达到各年龄段推荐摄入量范围即可。通常饮食中的钙摄入即可满足钙需求。对于佝偻病严重、钙摄入不足的患者可以考虑短期补充钙剂(3个月左右)。不推荐给低血磷性佝偻病/骨软化症额外补充钙剂,因为会增加肾脏钙化和肾结石的风险[71,72,73,74]。HHRH的患者不建议额外补充钙剂。
(2)成年患者:对于无症状、无反复牙周炎和牙周脓肿和近期无骨骼或口腔科手术或经临床判断骨软化症活动度不高(血ALP不高)的成年低血磷性佝偻病/骨软化症患者,通常无需活性维生素D和中性磷等治疗。因为上述治疗除增加患者负担外,还可能出现继发性甲旁亢、肾脏钙化和结石的风险。
对于有症状(肌肉无力、骨骼和关节疼痛)、反复牙周炎和牙周脓肿、近期进行骨骼或口腔科手术或经临床判断骨软化症活动(血ALP升高)的成年低血磷性佝偻病/骨软化症的患者,建议使用活性维生素D和磷酸盐常规治疗。这些治疗可改善疼痛、减轻骨软化症和减少牙周炎和牙周脓肿发病频率。
成人低血磷性佝偻病/骨软化症建议使用骨化三醇0.25~0.75 μg/d或阿法骨化醇0.5~1.5 μg/d。磷酸盐补充剂的剂量应为750~1 600 mg/d(基于元素磷含量确定),分2~4次服用[48,52,54,59,60]。为避免胃肠道不良反应,应从小量开始,逐渐增加剂量。
普通维生素D的使用同一般人群相仿,需要注意纠正维生素D缺乏,将25OHD的水平维持在30 ng/mL以上。推荐成年低血磷性佝偻病/骨软化症保持正常钙摄入和低钠饮食,以减少尿钙排泄。
(3)妊娠及哺乳期:建议低血磷性骨软化症的妇女在妊娠和哺乳期接受常规的磷酸盐(中性磷)和活性维生素D治疗。之前未接受常规治疗者建议开始治疗,已经接受治疗者除注意调整剂量,孕期和哺乳期所需的磷酸盐的剂量可能需要适当增加,最高可达2 000 mg/d。孕期和哺乳期患者治疗中应进行较密切的生化指标监测[75]。
(4)常规治疗的不良反应:口服磷酸盐的常见不良反应有腹痛和腹泻,从小剂量开始逐渐滴定增加剂量有助于减少或避免胃肠道反应。长期口服磷酸盐治疗可刺激PTH的分泌,导致继发性甲旁亢,甚至发生三发性甲旁亢的可能。为避免长期治疗继发性甲旁亢的发生,强调在治疗中需要监测血PTH的水平,疗效的监测中不应以血磷的纠正为目标,及时加用或调整活性维生素D的剂量,否则容易导致中性磷过量和诱发继发性甲旁亢。一旦患者出现三发性甲状旁腺功能亢进,应考虑甲状旁腺切除。
长期补充磷酸盐和使用活性维生素D常规治疗可能引起肾钙质沉着或肾结石[3,22,33,66,67,68,69,70,76]。肾钙质沉着发病风险与口服磷酸盐日剂量呈正相关,而其与活性维生素D治疗剂量是否相关尚不明确[3,22,66,68,69,70,77]。治疗中需要监测24 h尿钙或次尿钙的浓度,尿钙水平应<4 mg/kg 24 h或尿钙/肌酐<0.3 mg/mg。每年监测肾功能和泌尿系超声。
国外研究显示,XLH患者终身高男性为(143.7±8.56)cm, Z-值-3.7±1.62,女性为(140.8±7.46)cm, Z-值-2.54±1.42,均显著低于正常人[21,22,23,25,33,55,62,78,79,80,81,82]。由于生长激素可以促进肾小管对磷的重吸收使血磷升高,但是长期治疗提升血磷的作用不显著,同时生长激素治疗可能促使生长速度提高,可作为低血磷性佝偻病/骨软化症的辅助治疗。但是是否将生长激素用于低血磷性佝偻病/骨软化症的治疗尚存在争议。其有益作用可能促使生长速度提升,改善患者的身高。不利作用包括可能使佝偻病加重、下肢畸形加重、不能改善终身高。因此并不将重组人生长激素(recombinant human growth hormone, rhGH)作为低血磷性佝偻病的常规治疗。但是对身高极低的患者(Z-值<-2.4)采用生长激素治疗可能获益更多[83,84]。
另外,有文献报道SLC34A3突变导致的HHRH类型,重组人生长激素似乎可使患者身高受益[85]。
(1)儿童患者:随着对低血磷性佝偻病认识的提高,多数患者可能得到早期诊断和规律治疗,下肢畸形的发生和严重程度均明显改善。多数患者可能无需骨科手术治疗,但还是推荐骨科医生早期参与多学科诊疗团队,决定是否对患者给予手术干预。骨科手术仅限于那些经过正规的药物治疗仍不能矫正畸形的患者。6岁以下的患者即使有较严重的下肢畸形,也建议先使用药物治疗,因为药物治疗可能使下肢畸形得以矫正。如果患者在停止生长后仍存在严重的下肢畸形可以考虑采用截骨正畸手术。部分医疗中心对处于生长期的患儿开展低创骨骺引导性生长手术,将8字板置于生长较快侧的骨骺生长板的上下端,抑制植入侧的生长,随着患儿的生长下肢骨骼畸形和下肢的长度逐渐矫正。但必须密切监测,及时取出内置8字板,防止矫枉过正。
建议至少药物治疗12个月以后再选择性行手术治疗,对于经过药物优化治疗后存在持续性畸形的,建议考虑手术治疗,严重的下肢畸形可以通过在畸形的部位进行截骨治疗,但截骨术并发症较多且容易复发畸形。不建议使用石膏治疗X-连锁低磷血症患儿的下肢畸形。在患儿早期可进行引导生长微创手术[86]。在手术中,将一个小的金属板放置在骨骺的骨内侧或外侧表面(用于治疗外翻或内翻畸形),然后在两侧放置一颗螺钉,最后可以使靠近骨骺的地方垂直生长,随着时间的推移,骨呈对称生长,可以慢慢矫正畸形[87,88]。引导生长术取决于儿童剩余的骨生长潜力,因此必须在骨骼发育成熟(女孩14岁和男孩16岁)前至少2~3年实施,当手术在儿童晚期或骨骼成熟后进行时,截骨术的并发症减少。
(2)成年患者:成年患者如果存在严重的下肢畸形或发生病理性骨折时建议手术,因为单纯药物治疗很难再使低血磷性骨软化症患者的骨骼畸形得以矫正。同时对于严重关节畸形、关节炎、或严重的附丽病导致关节功能障碍的患者推荐实施人工关节置换术。部分患者会出现严重的椎体功能障碍、椎管狭窄等,病情严重,影响患者生活质量者建议行手术治疗,以缓解相关的压迫症状。对于低血磷性骨软化症的成年患者在手术前后均建议继续或开始正规的药物治疗。如果之前没有接受药物治疗的患者建议在手术前至少3~6个月进行药物治疗,手术后继续药物治疗至少半年或至骨愈合。药物治疗会促进骨科手术愈合和防治人工关节松动。
(1)口腔健康:几乎所有低血磷性佝偻病/骨软化症患者均存在口腔健康问题,必须予以高度重视。低血磷性佝偻病/骨软化症患者存在牙本质和牙釉质的缺陷,导致口腔和牙髓腔的保护屏障缺陷,易于出现自发性牙髓感染。患儿表现为乳牙的萌出延迟,换牙时恒牙萌出延迟,牙齿拥挤,排列不整齐,反复多发的牙周脓肿或牙龈炎。成年患者会出现牙齿过早脱落,严重者全口牙齿脱落。牙X线片上可见乳牙或恒牙的牙髓腔增大,且牙髓角变长[54,89],牙周的硬骨板缺失,牙齿周围的牙槽骨往往减少[20,90]。
正规的药物治疗可能减少低血磷性佝偻病/骨软化症患者的口腔并发症。注意口腔卫生,建议每次餐后刷牙,每天刷牙2~3次。定期口腔检查,保持口腔卫生。牙周脓肿或牙龈炎发作时可使用抗菌素。必要时可以考虑拔牙或根管治疗。为了防止细菌通过牙釉质的微裂隙侵入牙本质和牙髓,建议对儿童低血磷性佝偻病患者进行乳牙和恒牙的咬合面封闭治疗。牙齿畸形的患儿进行正畸治疗时,必须注意口腔卫生并坚持针对低血磷性佝偻病的常规药物治疗。成年患者进行相关的口腔操作时如拔牙、植牙等,均建议坚持常规的药物治疗。
(2)听力减退和颅骨异常:低血磷性佝偻病/骨软化症的患者可能出现听力减退、发作性耳鸣、耳聋和眩晕[91,92]。可能与广泛性骨硬化、岩骨增厚以及中度内耳道(特别是中部)狭窄有关[91,92]。其治疗与其他原因导致的听力异常相似。
大约有三分之二的低血磷性佝偻病的患儿会发生颅缝早闭,但多数无症状也无需特殊处理。但极少部分患儿会表现出颅高压的症状,需要脑外科处理。约四分之一的低血磷性佝偻病/骨软化症患者进行MRI或CT检查时,会发现小脑扁桃体下疝畸形(Chiari畸形Ⅰ型),极少数患者可表现出下脑干和上颈髓压迫,和(或)导致脊髓空洞症的表现,需要神经外科处理。
(3)葡萄糖和脂代谢功能受损:低血磷性佝偻病/骨软化症的患者有可能伴有肥胖、葡萄糖和脂代谢功能受损[93,94,95,96]。其原因尚不清楚,可能与患者活动能力较低、运动减少有关[18,19,33,49,97,98]。儿童患者在发生骨折之后、正畸手术后不能正常活动,或肿瘤性骨软化症的患者发生骨折或活动受限,不能下床活动时容易出现肥胖、糖耐量受损甚至糖尿病。应积极治疗,尽早恢复患者的活动能力,控制饮食,减轻体重,必要时予以相应的降糖治疗。
近年来,一些治疗低血磷性佝偻病/骨软化症的新型药物正在不断被开发和用于临床。全人源性FGF23单克隆抗体——布罗索尤单抗(Burosumab)在继欧盟、美国、日本等上市用于低血磷性佝偻病/骨软化症的治疗后,该药于2021年1月已经经我国国家药品监督管理局批准用于治疗成人和1岁及以上儿童X-连锁低磷血症(XLH)的治疗,同年3月获批用于TIO的治疗。其他旨在抑制高水平FGF23发挥效应的药物亦正处于研发阶段,部分新药可能会在未来几年进入临床。例如,FGF23受体拮抗剂NVP-BGJ398目前已进入研发后期[99]。另外,研究还确定了多个其他潜在的药物治疗靶点,包括拮抗已知的可抑制骨矿化的多肽以及调节FGF23蛋白的表达[100,101]。以下就布罗索尤单抗治疗做如下推荐。
(1)儿童患者:布罗索尤单抗用于治疗≥1岁儿童的低血磷性佝偻病患者[102]。布罗索尤单抗治疗儿童低血磷性佝偻病的临床效果包括:血磷水平上升至年龄-相关正常值范围的下限以上;TmP/GFR显著升高,血1,25(OH)2D水平升高;佝偻病严重程度显著降低[基于佝偻病严重程度评分(rickets severity score, RSS)和放射学总体印象变化(radiographic global impression of change, RGI-C)评分结果判定];体能显著改善[基于6分钟步行试验(6-minute walk test, 6MWT)中的步行距离测定结果];患者报告疼痛和功能残疾[采用北美小儿骨科学会儿科结局数据收集量表(Pediatric Outcomes Data Collection Instrument, PODCI)测定]也显著减少。
布罗索尤单抗推荐的起始剂量方案是0.8 mg/kg,四舍五入至最接近的10 mg,每2周1次。最低起始剂量为10 mg[86]。
在开始本品治疗后,在治疗的前3个月每4周测定1次空腹血磷,此后酌情进行测定。如果血磷高于各年龄段的参考值下限且低于5 mg/dL(1.61 mmol/L),则继续使用相同剂量进行治疗。在剂量调整后4周重新评估空腹血磷水平。本品剂量调整间隔不应小于每4周1次。
如果血磷低于所在年龄段的参考值下限,则根据
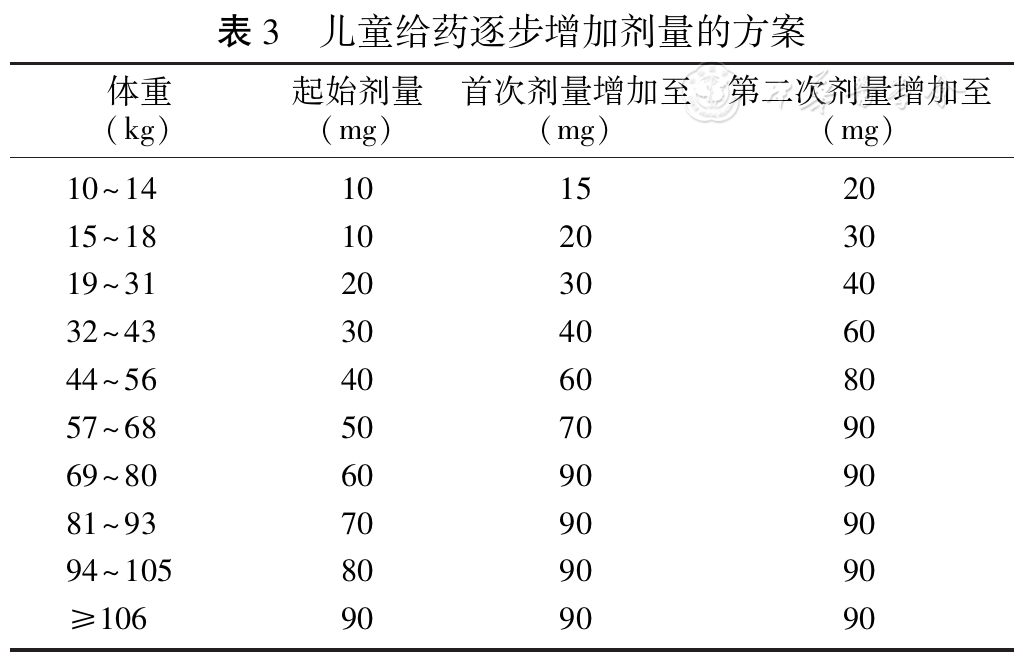
如果血磷高于5 mg/dL(1.61 mmol/L),则停用下一剂,并在4周内重新评估血磷水平。患者的血磷水平必须降至各年龄段的参考值下限,才可重新开始本品治疗。一旦血磷低于相应年龄段的参考值下限,则可按停药前大约一半的起始剂量(最大剂量为40 mg)重新开始治疗,每2周1次。在剂量调整后4周重新评估血磷水平。如果重新开始给药后血磷水平仍低于各年龄段的参考值下限,则可以根据附
(2)成年患者:布罗索尤单抗治疗成年低血磷性骨软化症-相关骨痛患者的结果表明布罗索尤单抗短期治疗(6~12个月)的结局如下[103]:可使TmP/GFR显著升高,血清磷酸盐水平上升至正常值范围,同时1,25(OH)2D水平升高;骨软化症治愈以及活动性骨折(active fractures)和假性骨折愈合加速;以及僵硬度显著减轻[采用加拿大西安大略省和麦克马斯特大学骨关节炎评分(Western Ontario and the McMaster Universities Osteoarthritis Index, WOMAC)僵硬度分量表测量]。但是与安慰剂治疗相比,WOMAC体能分量表和简明疼痛量表评分的减少并未见显著差异。
值得注意的是,成年低血磷性骨软化症患者大部分是轻症或无症状,以上临床研究结果主要针对中、重度骨痛的患者,这些研究结果可能难以外推至真实世界患者。
成人的推荐剂量方案为1 mg/kg,四舍五入至最接近的10 mg,每4周1次。在开始本品治疗后,在治疗的前3个月每月评估1次空腹血磷(在给药后2周测定),此后酌情进行测定。如果血磷在正常范围内,继续使用相同剂量进行治疗。如果血磷高于正常值上限,则停用下一剂,并在4周后重新评估血磷水平。患者的血磷必须低于正常值下限才能重新开始本品治疗。一旦血磷低于正常值下限,则可按停药前大约一半的起始剂量(最大剂量为40 mg)重新开始治疗,每4周1次。在剂量发生任何变化后2周重新评估血磷水平。本品剂量调整间隔不应小于每4周1次。
布罗索尤单抗最常见的不良反应是注射部位反应、头痛和四肢疼痛。应在布罗索尤单抗治疗开始前至少1周时停止常规治疗,以进行药物洗脱,并证明空腹血磷水平低于相应年龄的正常参考范围。
儿童和成人的药代动力学和药效学研究结果相似,药物半衰期约为19 d,布罗索尤单抗血清浓度达峰时间为注射后7~11 d,与血清磷酸盐水平和TmP/GFR升高时间平行,从而支持其药代动力学与药效学直接相关[104]。因此,建议监测剂量滴定期间2次注射给药时点间(理想时点是末次注射后7~11 d)的空腹血清磷酸盐水平,以避免高磷血症。稳定剂量治疗3个月后,建议监测注射给药前的磷酸盐水平,以检测有无低血磷症。布罗索尤单抗剂量的调整频率不应超过每4周1次,建议采取至少2个月的较长间隔进行剂量调整。如治疗开始前的磷酸盐水平在年龄-相关正常参考范围内或合并严重肾功能不全时(因为此类患者有发生高磷血症的危险),不得给予布罗索尤单抗治疗。应将空腹血磷水平目标设定为相应年龄的正常参考范围下限,以减少异位矿化的风险。
建议对低血磷性佝偻病/骨软化症的患者进行长期的诊疗,需要多学科团队参与的综合管理、评估和随访。由于低血磷性佝偻病/骨软化症患者的临床、生化和放射学特征、长期并发症在患者间存在很大差异,因此,最终应根据患者的临床特点、疾病发展阶段和对治疗的反应,并根据临床医生的专业判断对患者制定个体化的治疗和监测方案。
低血磷性佝偻病的患儿,在初诊和随诊中的临床评估应包括:身高、生长速度、体重、步态、活动能力、疼痛程度、骨骼畸形状况、口腔和牙齿、听力及相关神经系统的症状和体征等。
准确测量身高,评估矮小的程度,计算每年或每半年的生长速度。下肢畸形的程度与佝偻病的活动状态相关。建议测定并记录髁间距、踝间距、胫骨或股骨间距等[105,106]。单一临床测量并不能充分评估下肢畸形和关节对线情况,需借助放射学评估完成。对于严重肢体畸形患者,应由有代谢性骨病诊治经验的骨科医生完成评估。此类评估应包括肢体长度和对线(冠状平面和矢状面)以及下肢扭转特征评估。
记录患儿的步态。年龄在5岁以上的患儿,推荐每年1次的6MWT评估患儿的活动能力[107]。
建议每年应完成2次以上口腔和牙齿检查,12岁左右完成正畸评估,之后逐渐扩展牙齿评估范围并过渡到成人检查诊疗。每次诊视时,都应记录牙脓肿的数量和急性口腔感染(包括颌面部蜂窝组织炎)的发作次数(因为这些是牙齿矿化受损的间接指标)。
对于年龄小于5岁伴头围增加不足、头部形状异常或有神经系统体征(包括颅内压升高引起的头痛和呕吐)的儿童,应考虑颅缝早闭[108,109,110]。临床检查中,应评估有无脊柱前凸、后凸和(或)侧凸。
低血磷性骨软化症的成年患者的随诊需要根据患者的病情和治疗状况作出临床随访计划。重点评估患者的疼痛、活动能力、骨骼畸形或骨折情况等。
低血磷性佝偻病/骨软化症患者的初诊和随访中,应包括以下生化指标:血钙、磷、ALP水平,25OHD水平、肌酐、PTH水平、尿钙与尿磷水平或排泄量,以及TmP/GFR。
需要指出空腹血磷水平不能作为传统的常规治疗的疗效检测指标或目标,但是对于接受布罗索尤单抗治疗患者,空腹血磷水平可作为评估疗效和调整剂量的指标。ALP水平是佝偻病/骨软化症病情活动度的可靠的生物标记物[25,58,59,60,111]。儿童中,骨特异性ALP约占血清总ALP的80%~90%,因此在儿童中可采用总ALP作为指标。成人中,由于约50%的循环ALP源自肝细胞,因此首选骨特异性ALP[112]。佝偻病或骨软化症未得到充分治疗时,可见ALP水平升高,尿钙水平通常降低。反之,佝偻病治愈后,ALP水平趋于正常,尿钙水平则开始增加[24,25]。口服磷酸盐补充剂可引起继发性甲状旁腺功能亢进,因此应定期测定PTH水平[61,76,113,114,115]。如PTH升高,则表明磷酸盐的用量偏大或活性维生素D剂量不足。需测定血清和尿钙水平,以评估活性维生素D治疗的安全性。对于接受治疗的患者,因血清FGF23水平并不能指导治疗,故不推荐定期测定[30,32,116,117]。
布罗索尤单抗治疗期间,血清1,25(OH)2D水平可能升高;因此,建议每6个月测量1次血清1,25(OH)2D水平,并结合尿钙排泄水平,作为安全性参数进行综合分析[50,118,119,120]。
对低血磷性佝偻病的影像学评估最常用的是双手腕关节正位和双侧膝关节X线摄影。据此可以判断佝偻病是否存在,并对佝偻病的严重程度进行评分,前后比较和随访,建议治疗开始阶段每3~6个月,之后每1~2年检查1次。其他骨骼X线摄影可以根据骨骼畸形、骨痛等情况进行检查。全身骨显像有助于了解骨骼病变的范围和严重程度,但不作为常规随访检查项目。成年低血磷性骨软化症的患者随访常根据骨痛的部位展开检查,常用检查部位包括骨盆正位、胸腰椎正侧位、或可疑骨折及假骨折的部位。对怀疑患有严重关节病变或附丽病的患者,可随访检查双足、双踝关节、双膝关节、髋关节、颈椎等部位的X线影像。
建议对5岁儿童及近期有口腔症状的成人完善全口曲面断层片(上下颌和牙齿的X线摄影)检查,并应根据临床需要进行复查。
对于有症状的成人和儿童(例如持续头痛、呕吐或颅骨形状异常者),推荐完善头颅和(或)脊椎MRI评估,以排除颅缝早闭、Ⅰ型Chiari畸形或脊髓空洞症。为了避免X线摄影或计算机体层成像扫描(CT)造成过量辐射,可利用MRI的"黑骨(black bone)"序列完成颅骨成像检查,该技术可提供骨骼与其他组织的高对比度图像[121]。
每年进行肾脏超声检查是筛查肾钙沉积的首选方法[66,67,68,69,114]。
外周骨定量CT(peripheral quantitative CT, pQCT)或双能X线吸收法(dual energy X-ray absorptiometry, DXA)测量的骨密度对低血磷性佝偻病患者的临床价值有限[71,72,122]。这两种方法都无法诊断骨软化症。有趣的是,甚至有研究发现,儿科低血磷性佝偻病患者的松质骨体积骨密度升高[71,72,122],成年低血磷性骨软化症的患者腰椎骨密度也会增高,而这有可能是对长期骨骼矿化缺陷的代偿机制。
有部分评估低血磷性佝偻病/骨软化症患者心血管系统功能的研究报告了动脉压升高、左室肥厚和(或)病理性心电图等表现,但并非所有此类研究都反映上述结论[97,123,124,125,126,127,128]。因此,我们建议血压测量应至少每年1次,只有在血压持续升高的情况下,方可进行更为详细的检查。
TIO患者的随访:对于肿瘤完全切除的患者,最初可以每半年复查生化指标,特别是血磷,后期可以每年复查1次,同时应用DXA测量骨密度。对于不能明确肿瘤部位或者不能完全切除的患者,应该每3~6个月复查生化指标,以调整药物剂量,预防不良反应[129]。
低血磷性佝偻病/骨软化症是一大类罕见的慢性代谢性骨病,给患者的生长、活动能力和生活质量均造成巨大的损害。但由于该病相对罕见,造成公众,甚至是专业医师对该病的认识和重视程度均不高,以致许多患者不能得到及时诊断和长期正规的治疗。为此,中华医学会内分泌学分会与骨质疏松和骨矿盐疾病分会联合国内众多专家制订此指南,希望国内同行以此为依托,各中心组成以内分泌科、儿科、肾内科、骨科、放射科、口腔科和理疗科的专业医师的多学科团队,联系相关社会组织、志愿者和患者群体代表,协同工作、积极科普推广,提高对本病的认识,形成我国低血磷性佝偻病/骨软化症诊疗体系,对低血磷性佝偻病/骨软化症进行全方位和全生命周期的医疗照护。对此类患者进行精准的临床诊断,制定个体化的治疗方案,在传统治疗的基础上,不断探索新的、安全有效的治疗方法,不断积累经验提高我国低血磷性佝偻病/骨软化症的诊疗水平。随着科技的进步,特别是分子生物学技术的迅猛发展,相信在未来会有更多更好的方法用于此类疾病的诊断和治疗,并且可以期待在不久的将来此类疾病可能得到完全的预防或根治。
毕宇芳(上海交通大学医学院附属瑞金医院)、陈兵(陆军军医大学第一附属医院)、陈德才(四川大学华西医院)、陈刚(福建省立医院)、陈宏(南方医科大学珠江医院)、陈莉明(天津医科大学代谢病医院)、陈璐璐(华中科技大学同济医学院附属协和医院)、冯波(上海东方医院)、高政南(大连市中心医院)、谷卫(浙江大学医学院附属第二医院)、谷伟军(解放军总医院)、郭立新(北京医院)、洪天配(北京大学第三医院)、侯建明(福建省立医院)、侯新国(山东大学齐鲁医院)、姬秋和(第四军医大学西京医院)、焦凯(第四军医大学唐都医院)、金小岚(成都军区总医院)、匡洪宇(哈尔滨医科大学附属第一医院)、李成江(浙江大学医学院附属第一医院)、李静(中国医科大学附属第一医院)、李梅(北京协和医院)、李强(哈尔滨医科大学附属第二医院)、李小英(复旦大学中山医院)、李延兵(中山大学附属第一医院)、李艳波(哈尔滨医科大学附属一院)、李玉坤(河北医科大学第三医院)、李玉秀(中国医学科学院北京协和医院)、廖二元(中南大学湘雅二医院)、林华(南京大学医学院附属鼓楼医院)、刘超(南京中医药大学附属中西医结合医院)、刘建民(上海交通大学医学院附属瑞金医院)、刘建英(南昌大学第一附属医院)、刘礼斌(福建医科大学附属协和医院)、刘铭(天津医科大学总医院)、刘伟(上海交通大学医学院附属仁济医院)、陆志强(复旦大学附属中山医院)、罗玮(青海省人民医院)、吕朝晖(中国人民解放军总医院)、吕雪梅(西藏自治区人民医院)、母义明(中国人民解放军总医院)、宁光(上海交通大学医学院附属瑞金医院)、彭永德(上海交通大学附属第一人民医院)、乔虹(哈尔滨医科大学附属第二医院)、秦贵军(郑州大学第一附属医院)、秦映芬(广西医科大学第一附属医院)、曲伸(上海市第十人民医院)、全会标(海南省人民医院)、单忠艳(中国医科大学附属第一医院)、盛志峰(中南大学湘雅二医院)、石勇铨(海军军医大学附属长征医院)、时立新(贵州医科大学附属医院)、苏本利(大连医科大学附属第二医院)、苏恒(云南省第一人民医院)、孙子林(东南大学附属中大医院)、汤旭磊(兰州大学第一医院)、童南伟(四川大学华西医院)、王广(首都医科大学朝阳医院)、王桂侠(吉林大学白求恩第一医院)、王卫庆(上海交通大学医学院附属瑞金医院)、王新玲(新疆维吾尔自治区人民医院)、王颜刚(青岛大学附属医院)、王养维(陕西省人民医院)、翁建平(中国科学技术大学附属第一医院)、吴文(广东省人民医院)、夏维波(北京协和医院)、肖建中(北京清华长庚医院)、谢忠建(中南大学湘雅二医院)、徐进(山东省立医院)、闫朝丽(内蒙古医科大学附属医院)、严励(中山大学孙逸仙纪念医院)、杨刚毅(重庆医科大学附属第二医院)、杨静(山西医科大学第一医院)、杨涛(南京医科大学第一附属医院)、余学锋(华中科技大学同济医学院附属同济医院)、袁慧娟(河南省人民医院)、曾天舒(华中科技大学协和医院)、张波(北京中日友好医院)、张俊清(北京大学第一医院)、张力辉(河北医科大学第二医院)、张巧(贵州医科大学附属医院)、章振林(上海交通大学附属第六人民医院)、赵家军(山东省立医院)、郑宏庭(陆军军医大学第二附属医院)、周翔海(北京大学人民医院)、周智广(中南大学湘雅二院)、朱大龙(南京大学医学院附属鼓楼医院)、朱梅(天津医科大学总医院)
[1] Beck-Nielsen SS, Brock-Jacobsen B, Gram J, et al. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark[J]. Eur J Endocrinol, 2009,160(3):491-497.
[2] Endo I, Fukumoto S, Ozono K, et al. Nationwide survey of fibroblast growth factor 23(FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment[J]. Endocr J, 2015,62(9):811-816.
[3] Rafaelsen S, Johansson S, R?der H, et al. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, pheno-types, and treatment complications[J]. Eur J Endocrinol, 2016,174(2):125-136.
[4] Martin A, David V, Laurence JS, et al. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides(minhibins): ASARM-peptide(s)are directly responsible for defective mineralization in HYP[J]. Endocrinology, 2008,149(4):1757-1772.
[5] Feng JQ, Clinkenbeard EL, Yuan B, et al. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophy-siology of the heritable disorders of rickets and osteomalacia[J]. Bone, 2013,54(2):213-221.
[6] A gene(PEX)with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium[J]. Nat Genet, 1995,11(2):130-136.
[7] Liu S, Guo R, Quarles LD. Cloning and characterization of the proximal murine Phex promoter[J]. Endocrinology, 2001,142(9):3987-3995.
[8] Yuan B, Takaiwa M, Clemens TL, et al. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia[J]. J Clin Invest, 2008,118(2):722-734.
[9] Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis[J]. J Endocrinol, 2007,194(1):1-10.
[10] Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23[J]. Nat Genet, 2000,26(3):345-348.
[11] Feng JQ, Ward LM, Liu S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism[J]. Nat Genet, 2006,38(11):1310-1315.
[12] Levy-Litan V, Hershkovitz E, Avizov L, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene[J]. Am J Hum Genet, 2010,86(2):273-278.
[13] Rafaelsen SH, Raeder H, Fagerheim AK, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification[J]. J Bone Miner Res, 2013,28(6):1378-1385.
[14] Fukumoto S. FGF23-related hypophosphatemic rickets/osteomalacia: diagnosis and new treatment[J]. J Mol Endocrinol, 2021,66(2):R57-R65.
[15] Tieder M, Modai D, Samuel R, et al. Hereditary hypophosphatemic rickets with hypercalciuria[J]. N Engl J Med, 1985,312(10):611-617.
[16] Chesher D, Oddy M, Darbar U, et al. Outcome of adult patients with X-linked hypophosphatemia caused by PHEX gene mutations[J]. J Inherit Metab Dis, 2018,41(5):865-876.
[17] Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypopho-sphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children[J]. J Clin Endocrinol Metab, 1996,81(11):4075-4080.
[18] Beck-Nielsen SS, Brusgaard K, Rasmussen LM, et al. Phenotype presentation of hypophosphatemic rickets in adults[J]. Calcif Tissue Int, 2010,87(2):108-119.
[19] Che H, Roux C, Etcheto A, et al. Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms[J]. Eur J Endoc-rinol, 2016,174(3):325-333.
[20] Biosse Duplan M, Coyac BR, Bardet C, et al. Phosphate and vitamin D prevent periodontitis in X-linked hypophosphatemia[J]. J Dent Res, 2017,96(4):388-395.
[21] Sochett E, Doria AS, Henriques F, et al. Growth and metabolic control during puberty in girls with X-linked hypophosphataemic rickets[J]. Horm Res, 2004,61(5):252-256.
[22] Verge CF, Lam A, Simpson JM, et al. Effects of therapy in X-linked hypophosphatemic rickets[J]. N Engl J Med, 1991,325(26):1843-1848.
[23] Zivi?njak M, Schnabel D, Billing H, et al. Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets[J]. Pediatr Nephrol, 2011,26(2):223-231.
[24] Carpenter TO, Imel EA, Holm IA, et al. A clinician′s guide to X-linked hypophosphatemia[J]. J Bone Miner Res, 2011,26(7):1381-1388.
[25] Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood[J]. Endocr Connect, 2014,3(1):R13-R30.
[26] Penido MG, Alon US. Hypophosphatemic rickets due to perturbations in renal tubular function[J]. Pediatr Nephrol, 2014,29(3):361-373.
[27] Carpenter TO, Shaw NJ, Portale AA, et al. Rickets[J]. Nat Rev Dis Primers, 2017,3:17101.
[28] Brodehl J, Krause A, Hoyer PF. Assessment of maximal tubular phosphate reabsorption: comparison of direct measurement with the nomogram of Bijvoet[J]. Pediatr Nephrol, 1988,2(2):183-189.
[29] Yamazaki Y, Okazaki R, Shibata M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia[J]. J Clin Endocrinol Metab, 2002,87(11):4957-4960.
[30] Endo I, Fukumoto S, Ozono K, et al. Clinical usefulness of measurement of fibroblast growth factor 23(FGF23)in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement[J]. Bone, 2008,42(6):1235-1239.
[31] Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia[J]. N Engl J Med, 2003,348(17):1656-1663.
[32] Carpenter TO, Insogna KL, Zhang JH, et al. Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status[J]. J Clin Endocrinol Metab, 2010,95(11):E352-E357.
[33] Berndt M,Ehrich JH, Lazovic D, et al. Clinical course of hypophosphatemic rickets in 23 adults[J]. Clin Nephrol, 1996,45(1):33-41.
[34] Guven A, Al-Rijjal RA, BinEssa HA, et al. Mutational analysis of PHEX, FGF23 and CLCN5 in patients with hypophosphataemic rickets[J]. Clin Endocrinol(Oxf), 2017,87(1):103-112.
[35] Kinoshita Y, Saito T, Shimizu Y, et al. Mutational analysis of patients with FGF23-related hypophosphatemic rickets[J]. Eur J Endocrinol, 2012,167(2):165-172.
[36] Beck-Nielsen SS, Brixen K, Gram J, et al. Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and CLCN5 in patients with hypophosphatemic rickets[J]. J Hum Genet, 2012,57(7):453-458.
[37] Morey M, Castro-Feijóo L, Barreiro J, et al. Genetic diagnosis of X-linked dominant hypophosphatemic rickets in a cohort study: tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type[J]. BMC Med Genet, 2011,12:116.
[38] Ruppe MD, Brosnan PG, Au KS, et al. Mutational analysis of PHEX, FGF23 and DMP1 in a cohort of patients with hypophosphatemic rickets[J]. Clin Endocrinol(Oxf), 2011,74(3):312-318.
[39] Gaucher C, Walrant-Debray O, Nguyen TM, et al. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets[J]. Hum Genet, 2009,125(4):401-411.
[40] Ichikawa S, Traxler EA, Estwick SA, et al. Mutational survey of the PHEX gene in patients with X-linked hypophosphatemic rickets[J]. Bone, 2008,43(4):663-666.
[41] Holm IA, Nelson AE, Robinson BG, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets[J]. J Clin Endocrinol Metab, 2001,86(8):3889-3899.
[42] Tyynismaa H, Kaitila I, N?nt?-Salonen K, et al. Identification of fifteen novel PHEX gene mutations in Finnish patients with hypophosphatemic rickets[J]. Hum Mutat, 2000,15(4):383-384.
[43] Dixon PH, Christie PT, Wooding C,et al. Mutational analysis of PHEX gene in X-linked hypophosphatemia[J]. J Clin Endocrinol Metab, 1998,83(10):3615-3623.
[44] Holm IA, Huang X, Kunkel LM. Mutational analysis of the PEX gene in patients with X-linked hypophosphatemic rickets[J]. Am J Hum Genet, 1997,60(4):790-797.
[45] A gene(PEX)with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium[J]. Nat Genet, 1995,11(2):130-136.
[46] Goji K, Ozaki K, Sadewa AH, et al. Somatic and germline mosaicism for a mutation of the PHEX gene can lead to genetic transmission of X-linked hypophosphatemic rickets that mimics an autosomal dominant trait[J]. J Clin Endocrinol Metab, 2006,91(2):365-370.
[47] Christie PT, Harding B, Nesbit MA, et al. X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene[J]. J Clin Endocrinol Metab, 2001,86(8):3840-3844.
[48] Connor J, Olear EA, Insogna KL, et al. Conventional therapy in adults with X-linked hypophosphatemia: effects on enthesopathy and dental disease[J]. J Clin Endocrinol Metab, 2015,100(10):3625-3632.
[49] Econs MJ. Conventional therapy in adults with XLH improves dental manifestations, but not enthesopathy[J]. J Clin Endocrinol Metab, 2015,100(10):3622-3624.
[50] Carpenter TO, Imel EA, Ruppe MD, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia[J]. J Clin Invest, 2014,124(4):1587-1597.
[51] Ariceta G, Langman CB. Growth in X-linked hypophosphatemic rickets[J]. Eur J Pediatr, 2007,166(4):303-309.
[52] Baroncelli GI, Angiolini M, Ninni E, et al. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosph-atemic rickets[J]. Eur J Paediatr Dent, 2006,7(2):61-66.
[53] M?kitie O, Doria A, Kooh SW, et al. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets[J]. J Clin Endocrinol Metab, 2003,88(8):3591-3597.
[54] Chaussain-Miller C, Sinding C, Wolikow M, et al. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D[J]. J Pediatr, 2003,142(3):324-331.
[55] Balsan S, Tieder M. Linear growth in patients with hypophosphatemic vitamin D-resistant rickets: influence of treatment regimen and parental height[J]. J Pediatr, 1990,116(3):365-371.
[56] Bettinelli A, Bianchi ML, Mazzucchi E, et al. Acute effects of calcitriol and phosphate salts on mineral metabolism in children with hypophosphatemic rickets[J]. J Pediatr, 1991,118(3):372-376.
[57] Lempicki M, Rothenbuhler A, Merzoug V, et al. Magnetic resonance imaging features as surrogate markers of X-linked hypophosphatemic rickets activity[J]. Horm Res Paediatr, 2017,87(4):244-253.
[58] Tsuru N, Chan JC, Chinchilli VM. Renal hypophosphatemic rickets. Growth and mineral metabolism after treatment with calcitriol(1,25-dihydroxyvitamin D3)and phosphate supplementation[J]. Am J Dis Child, 1987,141(1):108-110.
[59] Chesney RW, Mazess RB, Rose P, et al. Long-term influence of calcitriol(1,25-dihydroxyvitamin D)and supplemental phosphate in X-linked hypophosphatemic rickets[J]. Pediatrics, 1983,71(4):559-567.
[60] Costa T, Marie PJ, Scriver CR, et al. X-linked hypophosphatemia: effect of calcitriol on renal handling of phosphate, serum phosphate, and bone mineralization[J]. J Clin Endocrinol Metab, 1981,52(3):463-472.
[61] Rasmussen H, Pechet M, Anast C, et al. Long-term treatment of familial hypophosphatemic rickets with oral phosphate and 1 alpha-hydroxyvitamin D3[J]. J Pediatr, 1981,99(1):16-25.
[62] Quinlan C, Guegan K, Offiah A, et al. Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment[J]. Pediatr Nephrol, 2012,27(4):581-588.
[63] Miyamoto J, Koto S, Hasegawa Y. Final height of Japanese patients with X-linked hypophosphatemic rickets: effect of vitamin D and phosphate therapy[J]. Endocr J, 2000,47(2):163-167.
[64] Harrell RM, Lyles KW, Harrelson JM, et al. Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. Induction and maintenance with phosphorus and calcitriol[J]. J Clin Invest, 1985,75(6):1858-1868.
[65] Glorieux FH, Marie PJ, Pettifor JM, et al. Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets[J]. N Engl J Med, 1980,303(18):1023-1031.
[66] Keskin M, Sava?-Erdeve ?, Sa?sak E, et al. Risk factors affecting the development of nephrocalcinosis, the most common complication of hypophosphatemic rickets[J]. J Pediatr Endocrinol Metab, 2015,28(11-12):1333-1337.
[67] Seikaly MG, Baum M. Thiazide diuretics arrest the progression of nephrocalcinosis in children with X-linked hypophosphatemia[J]. Pediatrics, 2001,108(1):E6.
[68] Eddy MC, McAlister WH, Whyte MP. X-linked hypophosphatemia: normal renal function despite medullary nephrocalcinosis 25 years after transient vitamin D2-induced renal azotemia[J]. Bone, 1997,21(6):515-520.
[69] Goodyer PR, Kronick JB, Jequier S, et al. Nephrocalcinosis and its relationship to treatment of hereditary rickets[J]. J Pediatr, 1987,111(5):700-704.
[70] Seikaly M, Browne R, Baum M. Nephrocalcinosis is associated with renal tubular acidosis in children with X-linked hypophosphatemia[J]. Pediatrics, 1996,97(1):91-93.
[71] Beck-Nielsen SS, Brixen K, Gram J, et al. High bone mineral apparent density in children with X-linked hypophosphatemia[J]. Osteoporos Int, 2013,24(8):2215-2221.
[72] Cheung M, Roschger P, Klaushofer K, et al. Cortical and trabecular bone density in X-linked hypophosphatemic rickets[J]. J Clin Endocrinol Metab, 2013,98(5):E954-E961.
[73] Oliveri MB, Cassinelli H, Bergadá C, et al. Bone mineral density of the spine and radius shaft in children with X-linked hypophosphatemic rickets(XLH)[J]. Bone Miner, 1991,12(2):91-100.
[74] Reid IR, Murphy WA, Hardy DC, et al. X-linked hypophosphatemia-skeletal mass in adults assessed by histomorphometry, computed-tomography, and absorptiometry[J]. Am J Med, 1991,90(1):63-69.
[75] Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation[J]. Endocr Rev, 1997,18(6):832-872.
[76] M?kitie O, Kooh SW, Sochett E. Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets[J]. Clin Endocrinol(Oxf), 2003,58(2):163-168.
[77] Vaisbich MH, Koch VH. Hypophosphatemic rickets: results of a long-term follow-up[J]. Pediatr Nephrol, 2006,21(2):230-234.
[78] Fuente R, Gil-Pe?a H, Claramunt-Taberner D, et al. X-linked hypophosphatemia and growth[J]. Rev Endocr Metab Disord, 2017,18(1):107-115.
[79] Capelli S, Donghi V, Maruca K, et al. Clinical and molecular heterogeneity in a large series of patients with hypophosphatemic rickets[J]. Bone, 2015,79:143-149.
[80] Borghi MM, Coates V, Omar HA. Evaluation of stature development during childhood and adolescence in individuals with familial hypophosphatemic rickets[J]. ScientificWorldJournal, 2005,5:868-873.
[81] Dudkiewicz I, Schindler A, Ganel A. Elongation of long bones for short stature in patients with hypophosphatemic rickets[J]. Isr Med Assoc J, 2003,5(1):66-67.
[82] Seikaly MG, Browne RH, Baum M. The effect of phosphate supplemen-tation on linear growth in children with X-linked hypophosphatemia[J]. Pediatrics, 1994,94(4):478-481.
[83] Meyerhoff N, Haffner D, Staude H, et al. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets[J]. Pediatr Nephrol, 2018,33(3):447-456.
[84] Baroncelli GI, Bertelloni S, Ceccarelli C, et al. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets[J]. J Pediatr, 2001,138(2):236-243.
[85] Dreimane D, Chen A, Bergwitz C. Description of a novel SLC34A3.c.671delT mutation causing hereditary hypophosphatemic rickets with hypercalciuria in two adolescent boys and response to recombinant human growth hormone[J]. Ther Adv Musculoskelet Dis, 2020,12:1759720X20912862.
[86] Haffner D, Emma F, Eastwood DM, et al. Clinical practice recommen-dations for the diagnosis and management of X-linked hypophosphataemia[J]. Nat Rev Nephrol, 2019,15(7):435-455.
[87] Gizard A, Rothenbuhler A, Pejin Z, et al. Outcomes of orthopedic surgery in a cohort of 49 patients with X-linked hypophosphatemic rickets(XLHR)[J]. Endocr Connect, 2017,6(8):566-573.
[88] Horn A, Wright J, Bockenhauer D, et al. The orthopaedic management of lower limb deformity in hypophosphataemic rickets[J]. J Child Orthop, 2017,11(4):298-305.
[89] Opsahl Vital S, Gaucher C, Bardet C, et al. Tooth dentin defects reflect genetic disorders affecting bone mineralization[J]. Bone, 2012,50(4):989-997.
[90] Beltes C, Zachou E. Endodontic management in a patient with vitamin D-resistant Rickets[J]. J Endod, 2012,38(2):255-258.
[91] O′Malley SP, Adams JE, Davies M, et al. The petrous temporal bone and deafness in X-linked hypophosphataemic osteomalacia[J]. Clin Radiol, 1988,39(5):528-530.
[92] Davies M, Kane R, Valentine J. Impaired hearing in X-linked hypophosphataemic(vitamin-D-resistant)osteomalacia[J]. Ann Intern Med, 1984,100(2):230-232.
[93] Zelenchuk LV, Hedge AM, Rowe PS. PHEX mimetic(SPR4-peptide)corrects and improves HYP and wild type mice energy-metabolism[J]. PLoS One, 2014,9(5):e97326.
[94] Angelin B, Larsson TE, Rudling M. Circulating fibroblast growth factors as metabolic regulators--a critical appraisal[J]. Cell Metab, 2012,16(6):693-705.
[95] Rowe PS. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway[J]. Crit Rev Eukaryot Gene Expr, 2012,22(1):61-86.
[96] Vaughn LK, Meyer RA
[97] Nehgme R, Fahey JT, Smith C, et al. Cardiovascular abnormalities in patients with X-linked hypophosphatemia[J]. J Clin Endocrinol Metab, 1997,82(8):2450-2454.
[98] Pronicka E, Popowska E, Rowińska E, et al. Anthropometric characteristics of X-linked hypophosphatemia[J]. Am J Med Genet A, 2004,126A(2):141-149.
[99] Javle M, Lowery M, Shroff RT, et al. Phase Ⅱ study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma[J]. J Clin Oncol, 2018,36(3):276-282.
[100] Fuente R, Gil-Pe?a H, Claramunt-Taberner D, et al. MAPK inhibition and growth hormone: a promising therapy in XLH[J]. FASEB J, 2019,33(7):8349-8362.
[101] Carpenter KA, Ross RD. Sclerostin antibody treatment increases bone mass and normalizes circulating phosphate levels in growing Hyp mice[J]. J Bone Miner Res, 2020,35(3):596-607.
[102] Imel EA, Glorieux FH, Whyte MP, et al. Burosumab versus conven-tional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial[J]. Lancet, 2019,393(10189):2416-2427.
[103] Insogna KL, Briot K, Imel EA, et al. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of Burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis[J]. J Bone Miner Res, 2018,33(8):1383-1393.
[104] Carpenter TO, Whyte MP, Imel EA, et al. Burosumab therapy in children with X-linked hypophosphatemia[J]. N Engl J Med, 2018,378(21):1987-1998.
[105] Yeo A, James K, Ramachandran M. Normal lower limb variants in children[J]. BMJ, 2015,350:h3394.
[106] Sass P, Hassan G. Lower extremity abnormalities in children[J]. Am Fam Physician, 2003,68(3):461-468.
[107] Saraff V, Schneider J, Colleselli V, et al. Sex-,age-,and height-specific reference curves for the 6-min walk test in healthy children and adolescents[J]. Eur J Pediatr, 2015,174(6):837-840.
[108] Rothenbuhler A, Fadel N, Debza Y, et al. High incidence of cranial synostosis and Chiari I malformation in children with X-linked hypophosphatemic rickets(XLHR)[J]. J Bone Miner Res, 2019,34(3):490-496.
[109] Vega RA, Opalak C, Harshbarger RJ, et al. Hypophosphatemic rickets and craniosynostosis: a multicenter case series[J]. J Neurosurg Pediatr, 2016,17(6):694-700.
[110] Murthy AS. X-linked hypophosphatemic rickets and craniosynostosis[J]. J Craniofac Surg, 2009,20(2):439-442.
[111] Ros I, Alvarez L, Gua?abens N, et al. Hypophosphatemic osteom-alacia: a report of five cases and evaluation of bone markers[J]. J Bone Miner Metab, 2005,23(3):266-269.
[112] Magnusson P, Sharp CA, Magnusson M, et al. Effect of chronic renal failure on bone turnover and bone alkaline phosphatase isoforms[J]. Kidney Int, 2001,60(1):257-265.
[113] Blydt-Hansen TD, Tenenhouse HS, Goodyer P. PHEX expression in parathyroid gland and parathyroid hormone dysregulation in X-linked hypophosphatemia[J]. Pediatr Nephrol, 1999,13(7):607-611.
[114] Alon U, Lovell HB, Donaldson DL. Nephrocalcinosis, hyperparath-yroidism, and renal failure in familial hypophosphatemic rickets[J]. Clin Pediatr(Phila), 1992,31(3):180-183.
[115] Schmitt CP, Mehls O. The enigma of hyperparathyroidism in hypophosphatemic rickets[J]. Pediatr Nephrol, 2004,19(5):473-477.
[116] Kubota T, Kitaoka T, Miura K, et al. Serum fibroblast growth factor 23 is a useful marker to distinguish vitamin D-deficient rickets from hypophosphatemic rickets[J]. Horm Res Paediatr, 2014,81(4):251-257.
[117] Igaki JM, Yamada M, Yamazaki Y, et al. High iFGF23 level despite hypophosphatemia is one of the clinical indicators to make diagnosis of XLH[J]. Endocr J, 2011,58(8):647-655.
[118] Ruppe MD, Zhang X, Imel EA, et al. Effect of four monthly doses of a human monoclonal anti-FGF23 antibody(KRN23)on quality of life in X-linked hypophosphatemia[J]. Bone Rep, 2016,5:158-162.
[119] Zhang X, Peyret T, Gosselin NH, et al. Population pharmacokinetic and pharmacodynamic analyses from a 4-month intradose escalation and its subsequent 12-month dose titration studies for a human monoclonal anti-FGF23 antibody(KRN23)in adults with X-linked hypophosphatemia[J]. J Clin Pharmacol, 2016,56(4):429-438.
[120] Zhang X, Imel EA, Ruppe MD, et al. Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody(KRN23)in the first multiple ascending-dose trial treating adults with X-linked hypophosphatemia[J]. J Clin Pharmacol, 2016,56(2):176-185.
[121] Dremmen M, Wagner MW, Bosemani T, et al. Does the addition of a " Black Bone" sequence to a fast multisequence trauma MR protocol allow MRI to replace CT after traumatic brain injury in children?[J]. AJNR Am J Neuroradiol, 2017,38(11):2187-2192.
[122] Veilleux LN, Cheung MS, Glorieux FH, et al. The muscle-bone relationship in X-linked hypophosphatemic rickets[J]. J Clin Endocrinol Metab, 2013,98(5):E990-E995.
[123] Takashi Y, Kinoshita Y, Hori M, et al. Patients with FGF23-related hypophosphatemic rickets/osteomalacia do not present with left ventricular hypertrophy[J]. Endocr Res, 2017,42(2):132-137.
[124] Nakamura Y, Takagi M, Takeda R, et al. Hypertension is a characteristic complication of X-linked hypophosphatemia[J]. Endocr J, 2017,64(3):283-289.
[125] Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy[J]. J Clin Invest, 2011,121(11):4393-4408.
[126] Alon US, Monzavi R, Lilien M, et al. Hypertension in hypopho-sphatemic rickets--role of secondary hyperparathyroidism[J]. Pediatr Nephrol, 2003,18(2):155-158.
[127] Moltz KC, Friedman AH, Nehgme RA, et al. Ectopic cardiac calcific-ation associated with hyperparathyroidism in a boy with hypophosp-hatemic rickets[J]. Curr Opin Pediatr, 2001,13(4):373-375.
[128] Vered I, Vered Z, Perez JE, et al. Normal left ventricular performance in children with X-linked hypophosphatemic rickets: a Doppler echocardiography study[J]. J Bone Miner Res, 1990,5(5):469-474.
[129] Jiang Y, Li X, Huo L, et al. Consensus on clinical management of tumor-induced osteomalacia[J]. Chin Med J(Engl), 2021,134(11):1264-1266.
