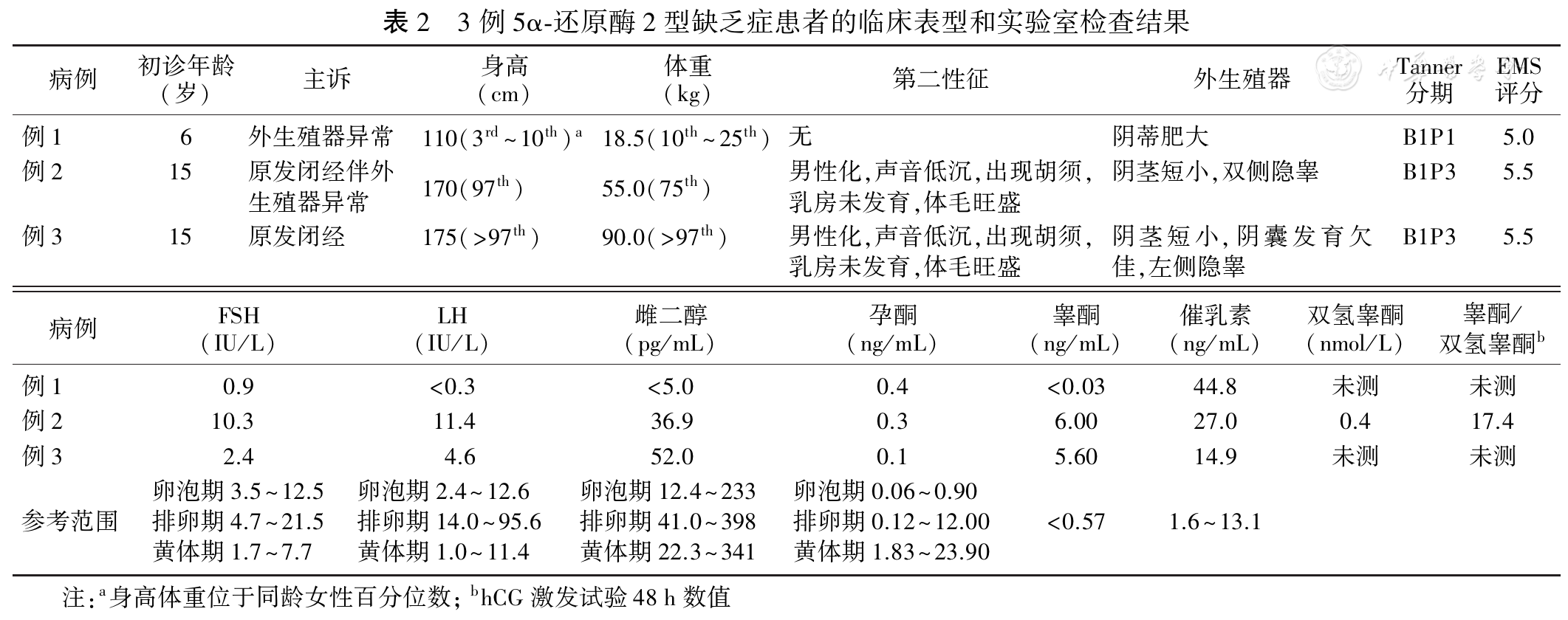
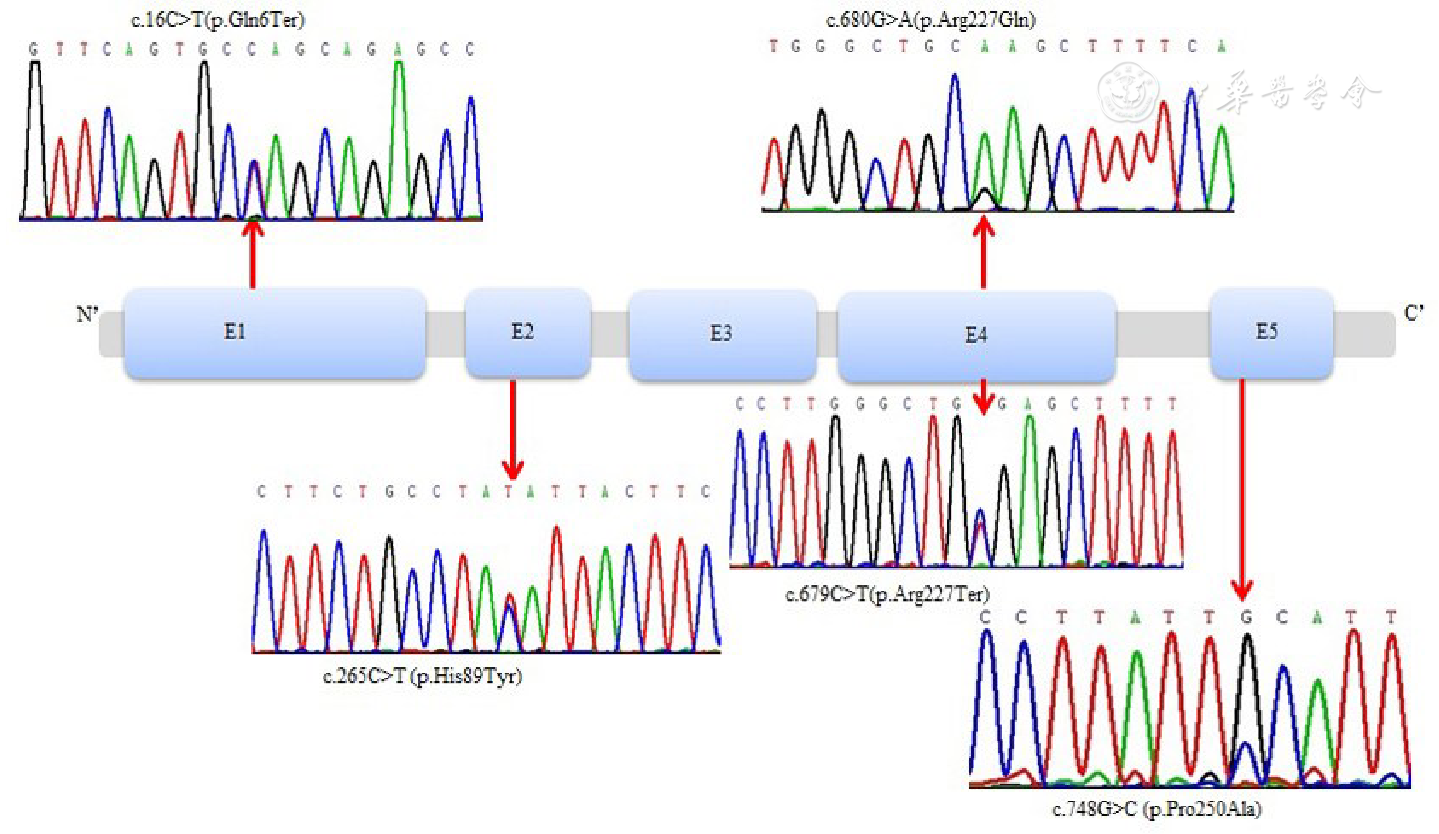
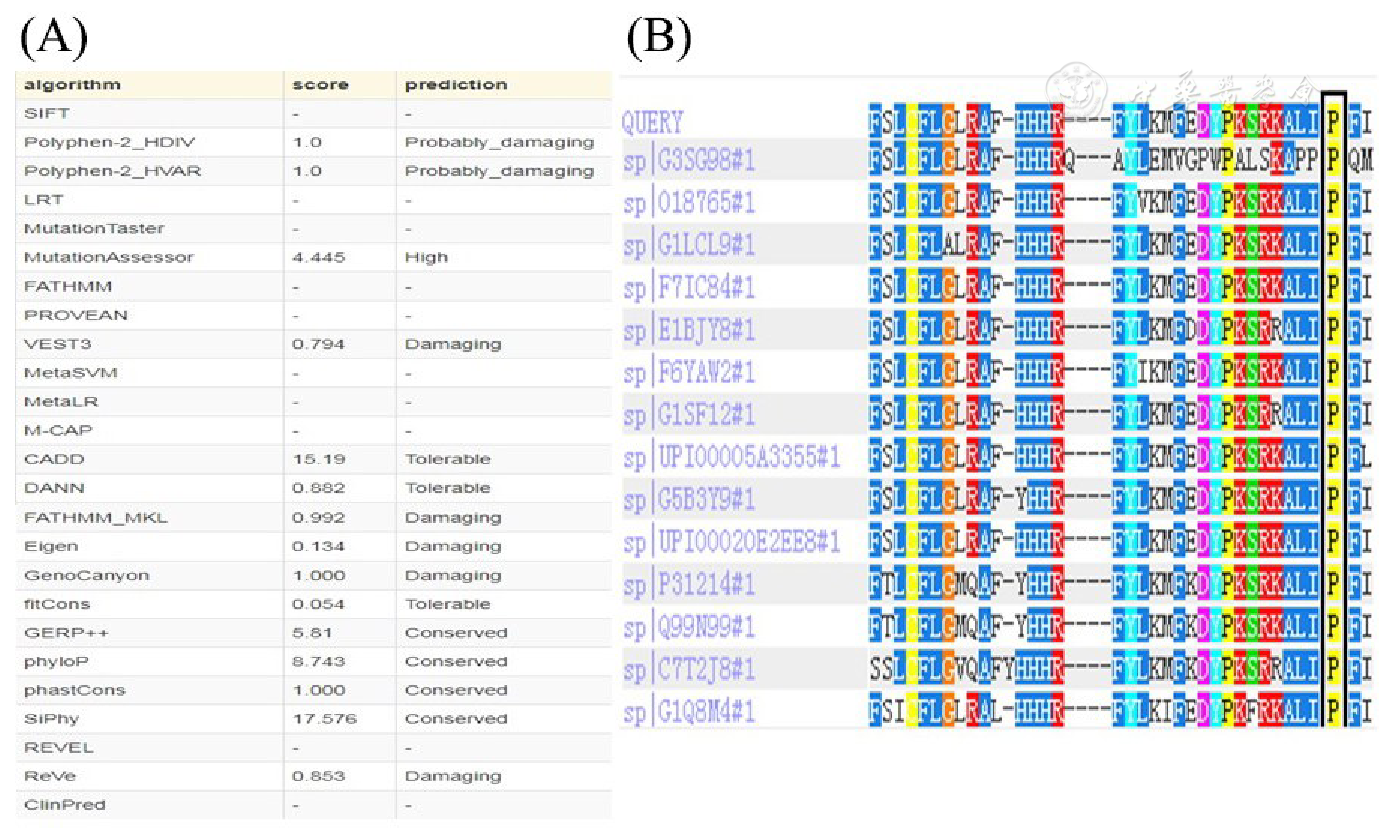
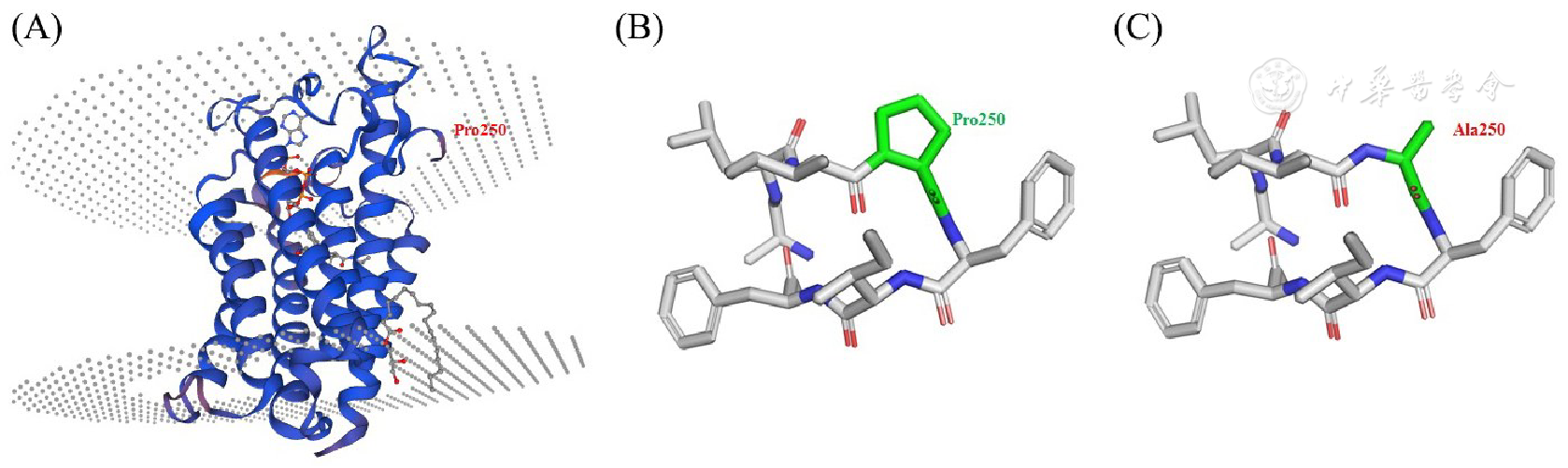
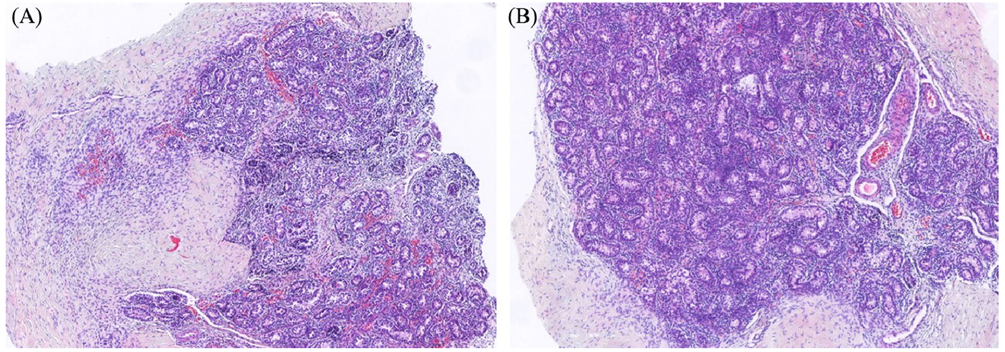
所有作者均声明不存在利益冲突参考文献
[1] Gui B, Song Y, Su Z, et al. New insights into 5α-reductase type 2 deficiency based on a multi-centre study: regional distribution and genotype-phenotype profiling of SRD5A2 in 190 Chinese patients[J]. J Med Genet, 2019,56(10):685-692. DOI: 10.1136/jmedgenet-2018-105915.
[2] Azzouni F, Godoy A, Li Y, et al. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases[J]. Adv Urol, 2012,2012:530121. DOI: 10.1155/2012/530121.
[3] 夏俊珂,罗晓,吴静,等. 五例Kallmann综合征患者的基因型-表型关系和遗传学研究[J]. 中华内分泌代谢杂志,2021,37(12):1106-1111. DOI: 10.3760/cma.j.cn311282-20210427-00261.
[4] Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations[J]. Nat Methods, 2010,7(4):248-249. DOI: 10.1038/nmeth0410-248.
[5] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015,17(5):405-424. DOI: 10.1038/gim.2015.30.
[6] Arnold K, Bordoli L, Kopp J, et al. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling[J]. Bioinformatics, 2006,22(2):195-201. DOI: 10.1093/bioinformatics/bti770.
[7] Sinnecker GH, Hiort O, Dibbelt L, et al. Phenotypic classification of male pseudohermaphroditism due to steroid 5 alpha-reductase 2 deficiency[J]. Am J Med Genet, 1996,63(1):223-230. DOI: 10.1002/(SICI)1096-8628(19960503)63:1<223::AID-AJMG39>3.0.CO;2-O.
[8] Yuan S, Meng L, Zhang Y, et al. Genotype-phenotype correlation and identification of two novel SRD5A2 mutations in 33 Chinese patients with hypospadias[J]. Steroids, 2017,125:61-66. DOI: 10.1016/j.steroids.2017.06.010.
[9] Shabir I, Khurana ML, Joseph AA, et al. Phenotype, genotype and gender identity in a large cohort of patients from India with 5α-reductase 2 deficiency[J]. Andrology, 2015,3(6):1132-1139. DOI: 10.1111/andr.12108.
[10] Avenda?o A, Paradisi I, Cammarata-Scalisi F, et al. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review[J]. Hormones(Athens), 2018,17(2):197-204. DOI: 10.1007/s42000-018-0013-9.
[11] 王瑞芳,董治亚,王伟,等. 52例尿道下裂患者5α-还原酶2型基因突变研究[J]. 中国实用儿科杂志,2012,27(9):693-696. DOI: CNKI:SUN:ZSEK.0.2012-09-020.
[12] 林胡,杨浩,傅君芬,等. 中国性发育异常患儿临床表型与基因型分析[J]. 中华儿科杂志,2022,60(5):435-441. DOI: 10.3760/cma.j.cn112140-20210927-00828.
[13] 温鹏强,王国兵,陈占玲,等. 41例5α-还原酶2型缺陷症患儿临床特点及SRD5A2基因突变分析[J]. 中华内分泌代谢杂志,2019(3):226-232. DOI: 10.3760/cma.j.issn.1000-6699.2019.03.008.
[14] Xia J, Wu J, Chen C, et al. Molecular study and genotype-phenotype in Chinese female patients with 46, XY disorders of sex development[J]. Gynecol Endocrinol, 2021,37(10):934-940. DOI: 10.1080/09513590.2021.1960307.
[15] 宋艳宁,范丽君,赵岫,等. 5α-还原酶缺乏症86例SRD5A2基因检测结果与临床表型分析[J]. 中华儿科杂志,2019,57(2):131-135. DOI: 10.3760/cma.j.issn.0578-1310.2019.02.013.
[16] 郑小琴,毛宇,邱碧原,等. 65例染色体核型为46,XY的尿道下裂患者AR和SRD5A2基因突变分析[J]. 第三军医大学学报,2018,40(11):998-1004. DOI: 10.16016/j.1000-5404.201712132.
[17] 袁诗敏,钟昌高,李秀蓉,等. 25例尿道下裂患者的核型及SRD5A2基因突变分析[J]. 中华医学遗传学杂志,2017,34(2). DOI: 10.3760/cma.j.issn.1003-9406.2017.02.001.
[18] Deeb A, Al Suwaidi H, Ibukunoluwa F, et al. Phenotype, Sex of Rearing, Gender Re-Assignment, and Response to Medical Treatment in Extended Family Members with a Novel Mutation in the SRD5A2 Gene[J]. J Clin Res Pediatr Endocrinol, 2016,8(2):236-240. DOI: 10.4274/jcrpe.2782.
[19] Fénichel P, Paris F, Philibert P, et al. Molecular diagnosis of 5α-reductase deficiency in 4 elite young female athletes through hormonal screening for hyperandrogenism[J]. J Clin Endocrinol Metab, 2013,98(6):E1055-E1059. DOI: 10.1210/jc.2012-3893.
[20] Cohen-Kettenis PT. Gender change in 46,XY persons with 5alpha-reductase-2 deficiency and 17beta-hydroxysteroid dehydrogenase-3 deficiency[J]. Arch Sex Behav, 2005,34(4):399-410. DOI: 10.1007/s10508-005-4339-4.
[21] Costa EM, Domenice S, Sircili MH, et al. DSD due to 5α-reductase 2 deficiency - from diagnosis to long term outcome[J]. Semin Reprod Med, 2012,30(5):427-431. DOI: 10.1055/s-0032-1324727.
[22] Matsubara K, Iwamoto H, Yoshida A, et al. Semen analysis and successful paternity by intracytoplasmic sperm injection in a man with steroid 5α-reductase-2 deficiency[J]. Fertil Steril, 2010,94(7):2770.e7-e10. DOI: 10.1016/j.fertnstert.2010.04.013.