纤维蛋白原(fibrinogen,Fg)又称为凝血因子Ⅰ,是一种相对分子质量为340 000的大分子糖蛋白,是血浆中含量最高的凝血因子,直接参与凝血过程的共同途径。遗传性异常纤维蛋白原血症(inherited dysfibrinogenemia)是一种纤维蛋白原基因缺陷引起Fg分子结构异常,从而导致其功能异常的遗传性疾病。该病较为罕见,且临床表现难以预测,可从无临床症状到危及患者生命,为一种通常由杂合突变或缺失突变引起的常染色体显性遗传病,少数为常染色体隐性遗传,可由于纤维蛋白肽释放异常、纤维蛋白单体聚合异常、纤维蛋白交联异常和纤维蛋白凝块纤溶异常等机制影响纤维蛋白原的正常功能[1]。临床表现为Fg活性降低,而Fg抗原大多正常,即Fg功能和抗原水平常不同步降低。国际上已报道的遗传性异常纤维蛋白血症家系有500多例(http://www.geht.org/),目前国内报道的遗传性异常纤维蛋白血症家系有40多例。
本研究中我们对1个遗传性异常纤维蛋白原血症家系进行临床表型和基因型分析,发现Fg γ链D区的一个尚未见报道的新突变,通过Fg功能相关试验、电镜观察纤维蛋白凝块等手段,探讨其发病机制,并对患者的临床出凝血预测进行探讨,有助于患者规避出凝血风险。
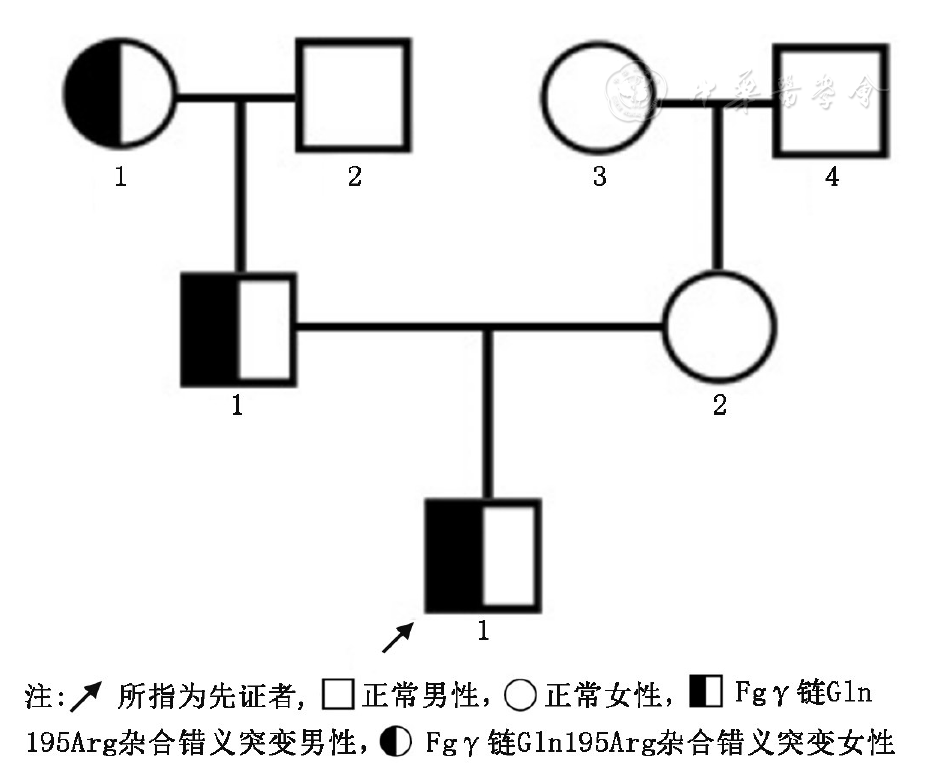
先证者,男,12岁,因右腹股沟斜疝欲手术治疗,术前检查发现活化部分凝血活酶时间(activated partial thromboplastin time, APTT)正常,凝血酶原时间(prothrombin time, PT)和凝血酶时间(thrombin time, TT)延长,Fg活性(fibrinogen activity, Fg︰C)明显降低。平素无出血或血栓病史,无肝炎、肾病史,无药物过敏史,无服用抗凝药物史。肝、肾功能正常。该家系无近亲婚配史,其他成员均无出血或血栓病史。家系图谱见图1,家系成员出凝血检测结果见表1。另外选取宁波华美医院102名健康体检者作为健康对照,用于排除基因多态性。本研究已经通过伦理委员会批准(PJ-NBEY-KY-2020-099-01),所有受试者均签署知情同意书。
在征得先证者及家系成员知情同意后,用枸橼酸钠抗凝管采集家系中3代7人外周血,2 000×g离心10 min后吸取上层血浆,分装,-80 ℃保存。日本Sysmex CA7000全自动凝血仪(美国Dade Berhing公司试剂盒)检测APTT、PT、TT。Fg︰C和纤维蛋白原抗原(fibrinogen antigen, Fg︰Ag)分别用凝固法(Clauss法)(德国Dade Berhing公司试剂盒,日本Sysmex CA7000全自动凝血仪)和免疫比浊法(上海太阳公司试剂盒,美国Beckman & Coulter公司Cx7全自动生化仪)进行检测。爬虫酶时间(reptilase time, RT)用磁珠法(法国Stago Reptilase试剂盒,法国Stago半自动血凝分析仪)检测。蛋白C活性(PC∶A)、蛋白S活性(PS∶A)和抗凝血酶活性(AT∶A)用发色底物法(德国Dade Behring公司试剂盒,日本Sysmex公司CA7000全自动血凝仪)测定。所有操作步骤均严格按照仪器和试剂盒说明书进行。
用血液基因组DNA提取试剂盒(北京Tiangen公司)抽提7名家系成员和102名健康对照者的外周血基因组DNA。参照文献[2]合成Fg扩增引物,扩增FGA、FGB和FGG基因的所有外显子及其侧翼序列。PCR反应体系为25 μl,10×含Mg2+缓冲液2.5 μl,dNTP 250 μmoL/L,上下游引物各0.5 μmoL/L,DNA模板250 ng,Taq DNA聚合酶1.5U(上海申能博彩公司产品)。PCR条件:95 ℃预变性5 min,95 ℃ 30 s、56 ℃ 30 s、72 ℃ 30 s,共30个循环,最后72 ℃延伸10 min[3]。PCR产物经琼脂糖凝胶电泳、割胶后,用琼脂糖DNA纯化试剂(上海申能博彩公司产品)回收并纯化PCR产物,末端标记双脱氧法进行测序(美国ABI 3700基因测序仪)。用Chromas软件将测序结果与GenBank中Fg的基因序列(M64982、M64983和M10014)进行比对。发现基因突变后,正向和反向重复测序3次进行确认。家系成员DNA则仅在先证者突变区域扩增测序。对尚未有报道的突变,在102名健康对照者中进行相应区域的扩增筛选,以排除多态性改变的可能。
先证者、其父亲、其祖母及健康对照血浆用凝胶加样缓冲液(含还原剂β-巯基乙醇)以1∶200稀释,100 ℃加热变性5 min,十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis)和转膜参照文献[4],使用的一抗为多克隆兔抗人纤维蛋白原抗体(丹麦DakoCytomation公司产品),二抗为HRP标记的山羊抗兔IgG抗体(Cell Signaling公司)。转膜后使用ECL化学发光检测试剂盒(Pierce公司)进行显影检测。
取20名凝血功能正常的健康体检者的混合血浆作为健康对照,取先证者及健康对照者枸橼酸钠抗凝血的乏血小板血浆,加入0.5 mol/L乙二胺四乙酸二钾至终浓度为10 mmol/L,混匀后再加入人凝血酶(上海莱士血液制品股份有限公司产品)至终浓度为20 NIH U/ml,立即混匀。37 ℃孵育60 min后,凝块形成,用50 mmol/L pH7.4三羟甲基氨基甲烷[Tris(hydroxymethyl)aminomethane]缓冲液洗5次后,加入血浆等体积的5 mol/L尿素溶液溶解凝块,用紫外分光光度计测蛋白浓度(280 nm),并按照以下公式计算Fg凝固能力:
向96孔板孔内加入91 μl血浆和9 μl 10 NIH U/ml人凝血酶,终浓度为0.9 NIH U/ml,立即用美国BIO-TEK微孔板分光光度计及KC4软件系统进行37 ℃ 365 nm连续动态读数,读数间隔时间为23 s。
向96孔板内加入40 μl先证者的乏血小板血浆及10 μl 10 NIH U/ml人凝血酶,终浓度为2 NIH U/ml,混匀后室温孵育3 h,凝块形成,用蒸馏水漂洗10次,加入2.5%戊二醛4 ℃固定过夜[4]。取健康对照者乏血小板血浆,作相同处理。将凝块标本送至电镜室,继续锇酸固定、乙醇梯度脱水、CO2临界点干燥、喷金镀膜等处理后,用PhilipsXL-30型扫描电镜在5 000×放大倍数下扫描观察并拍照。应用imageJ软件测量凝块电镜扫描照片中纤丝的直径,分别测量先证者和健康对照者的纤维蛋白凝块的100条纤丝的直径,用t检验比较两组直径数据,P<0.05为差异有统计学意义。
先证者APTT正常,PT和TT轻度延长,RT明显延长,Fg︰Ag轻度降低,Fg︰C明显降低。其父亲和祖母的APTT、PT和Fg︰Ag正常,TT轻度延长,RT明显延长,Fg︰C明显降低。家系其他成员所测指标均正常。具体检测结果见表1。家系成员PC︰A、PS︰A和AT︰A皆正常。
与GenBank中FGG(M10014)基因序列进行比对,发现先证者纤维蛋白原FGG基因第6外显子的4774位发生了A>G杂合错义突变,见图2,使得密码子由CAG变为CGG,导致FgγD结构域的195位谷氨酰胺被替换为精氨酸(Gln195Arg);其父亲和祖母均携带g. 4774A>G杂合错义突变,家系其他成员均显示为野生型。家系成员的Fg其他外显子和侧翼序列的测序结果未见异常。针对先证者的突变位点,对102名健康对照者的相应位点进行测序,未见相同突变,排除了基因多态性的可能性。查阅文献和检索Fg基因突变数据库(http://site.geht.org/base-de-donnees-fibrinogene/#)提示,Fg γ链Gln195Arg突变尚未见报道。
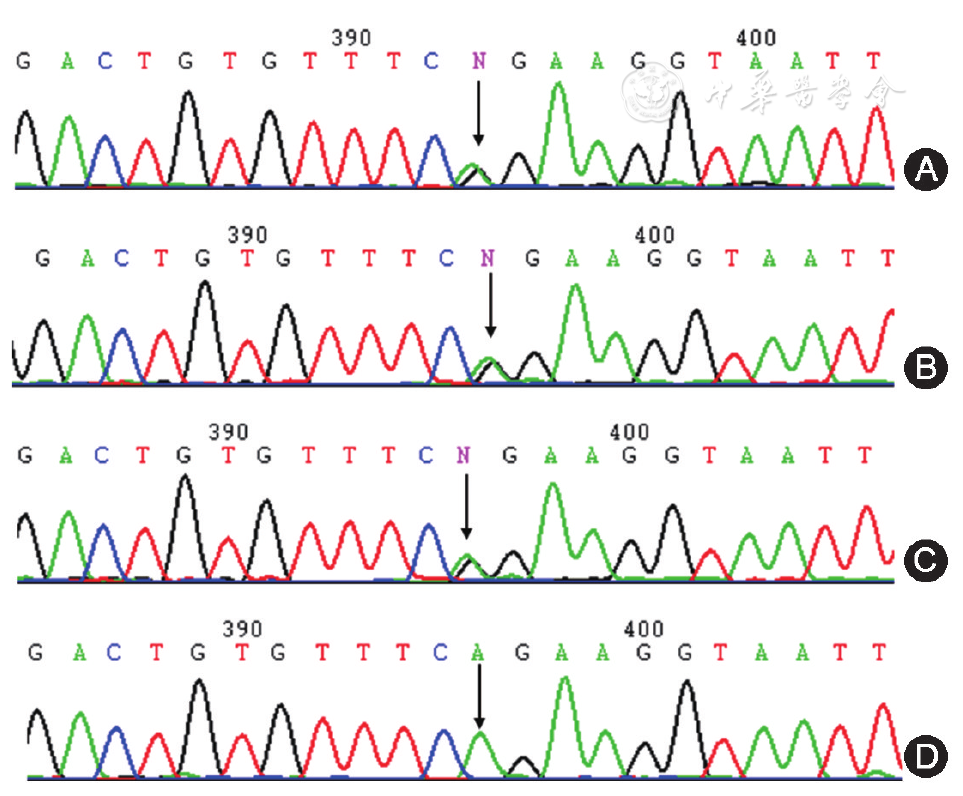
注:图A为先证者FGG基因第6外显子的正向测序图,箭头所指为g. 4774A>G(Gln195Arg)杂合错义突变;图B为先证者父亲FGG基因第6外显子的正向测序图,箭头所指为g. 4774A>G (Gln195Arg)杂合错义突变;图C为先证者祖母FGG基因第6外显子的正向测序图,箭头所指为g. 4774A>G(Gln195Arg)杂合错义突变;图D为健康对照者FGG基因第6外显子的正向测序图,箭头所指为4774位碱基,为野生型
与健康对照血浆相比,先证者及其父亲和祖母的血浆Fg的Aα、Bβ和γ条带分子量均无异常,结果见图3。
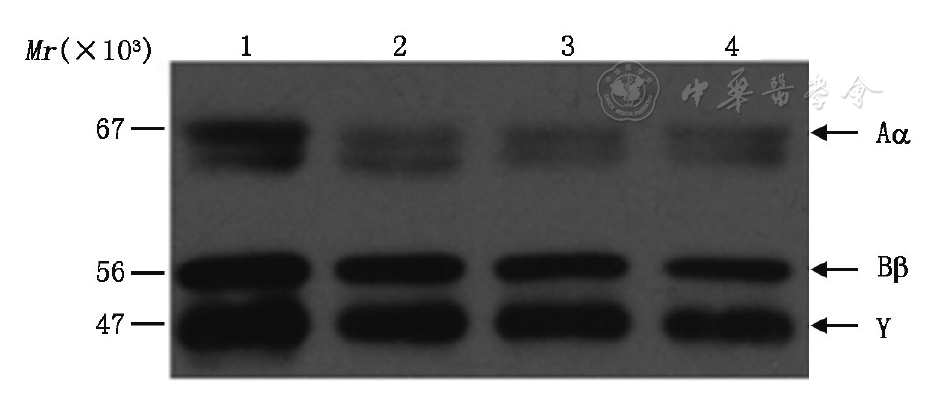
注:1为健康对照;2为先证者祖母;3为先证者父亲;4为先证者
先证者血浆的纤维蛋白原凝固率为49.3%,健康对照者血浆的纤维蛋白原凝固率为98.9%。
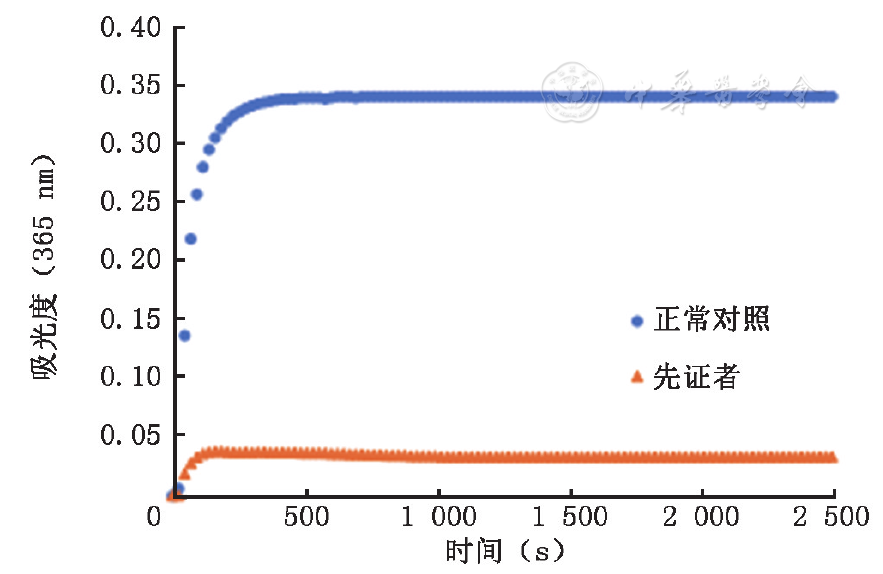
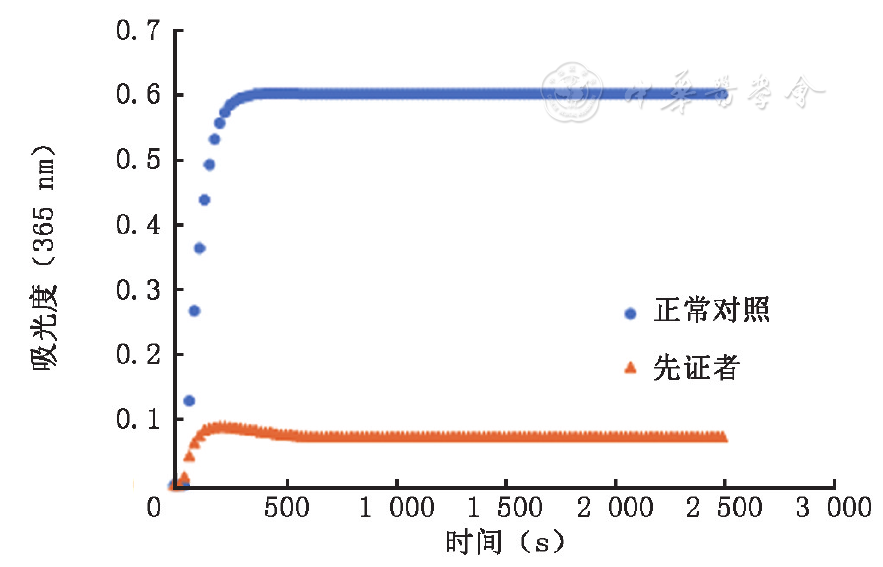
与健康对照者血浆的对应曲线相比较,先证者血浆的凝血酶或爬虫酶诱导的纤维蛋白聚集曲线的聚集起始时间均未见明显变化,但最大聚集率(峰值)均明显降低,聚集曲线均严重受损,提示先证者的纤维蛋白聚集功能活性下降,存在缺陷,见图4、图5。
测量凝块扫描电镜照片中100条先证者血浆Fg形成的纤维蛋白凝块的纤丝,直径为(194±48)nm(n=100),大于健康对照者凝块的纤丝直径(108±21)nm(n=100)(P<0.001);与健康对照者纤维蛋白凝块相比,先证者血浆Fg形成的纤维蛋白凝块的纤丝密度减小、排列稀疏,见图6。

注:图A为先证者血浆纤维蛋白凝块的扫描电镜照片;图B为正常人血浆纤维蛋白凝块的扫描电镜照片
Fg为由Aα、Bβ、γ 3条多肽链为组分、以链间二硫键即AαCys28、γCys8和Cys9相连构成的对称性二聚体(Aα、Bβ、γ)2,主要包括中央区(E区)和两个外围区(D区),其中D区由Bβ和γ链的羧基端(C-端)以及部分Aα链组成[5],包括α链111~197位氨基酸、β链134~461位氨基酸和γ链88~406位氨基酸。
遗传性异常纤维蛋白原血症最常见的突变为发生在Aα链的N-端区域或γ链的C-端区域的碱基置换突变,使得γ链的两个D区和E区之间、或者是两个D区之间的相互作用发生缺陷,从而导致早期凝块形成过程中纤维蛋白的装配发生缺陷[6]。γ链D区对Fg发挥正常功能至关重要,研究表明位于该结构域的Gly200Val[7]、Gln239His[8]、Arg275Cys[6]、Arg275His[6]、Ser313Ile[5]等氨基酸突变可引起不同程度Fg异常,导致异常纤维蛋白原血症或低纤维蛋白原血症。在本研究中,我们发现了一个由γ链D区Gln195Arg错义突变引起的遗传性异常纤维蛋白原血症家系,先证者的Fg︰Ag和Fg︰C分别为1.6 g/L和0.7 g/L,其父亲的Fg︰Ag和Fg︰C分别为2.0 g/L和1.0 g/L,其祖母的Fg︰Ag和Fg︰C分别为2.2 g/L和1.1 g/L。Westernblot检测结果显示,家系中三例患者的血浆Fg的Aα、Bβ和γ条带相对分子质量均无异常,均无异常条带出现。
该家系成员均无出血或血栓病史。遗传学分析发现,他们在FGG基因第6外显子的4774位存在A>G杂合突变,导致Gln195Arg错义突变,并排除了FGG基因多态性。查阅文献并检索纤维蛋白原基因突变数据库(http://site.geht.org/base-de-donnees-fibrinogene/#)确认,Gln195Arg为国际上未报道的新突变,该位点也未见其他突变类型的报道。
遗传性异常纤维蛋白原血症的临床表现差异极大,可从无临床表现到出血或血栓,很难预测。大部分的基因型和表型之间的关联至今还未明确阐明。纤维蛋白聚集能力及纤溶功能分析、纤维蛋白凝块网状结构分析等试验能够帮助预测遗传性异常纤维蛋白原血症的表型,但还未用于详细评估。有这类易致病的基因型的患者,在这种疾病的自然进程中也处于出血及或血栓风险,因此即使个体还没有出现出血或血栓症状、没有出凝血异常家族史,也应该被仔细评估和监测,尤其是要进行怀孕、分娩、外科手术的患者[6]。术前出凝血检查,并对异常情况进行进一步的研究分析,非常重要,这有利于临床医生进一步评估术中出血风险。
为了有助于预测该家系三位患者的表型,我们开展了Fg的功能和结构相关试验。实验结果表明,先证者血浆的Fg凝固率严重低下,约为健康对照者的一半,凝血酶或爬虫酶诱导的聚集曲线的光吸收度降低,提示其纤维蛋白聚集能力严重受损;用扫描电镜观察纤维蛋白凝块发现,患者凝块与健康对照者凝块相比存在明显异常,纤丝直径变粗,而凝块中纤丝密度变小、排列变稀疏,从而推测突变导致凝块比健康对照者凝块易渗透及穿透,从而不易网罗血小板及其他血细胞形成血凝块,导致止血功能下降;另外,纤丝密度变小、排列变稀疏很可能会引起纤维蛋白凝块的结构牢固度和稳定性减低,纤维蛋白凝块更容易被溶解,导致止血功能下降。通过以上实验结果进行预测,Gln195Arg突变导致的异常纤维蛋白原血症的患者可能存在出血风险。临床医生若为先证者行外科手术治疗腹股沟疝,要考虑到出血风险,并做好充分准备。
Fg γ链D区中Thr192-Ala286和Lys380-Leu392组成的结构域由7个反向平行β-片层、两个短螺旋结构和一个发夹结构排列而成[9],Gln195位于该结构域中。Gln为极性、中性氨基酸,相对分子量为128,而Arg为含碱性氨基酸,相对分子质量为157。Gln195Arg突变可能通过改变氨基酸的酸碱性、等电点、分子大小和疏水性等特性引起Fg γ链构象改变,从而影响整个蛋白分子的功能,导致遗传性异常纤维蛋白原血症。
综上所述,我们发现了一个由γ链D区Gln195Arg错义突变引起的遗传性异常纤维蛋白原血症家系,该突变尚未见报道。并通过Fg功能相关试验、扫描电镜观察纤维蛋白凝块等手段,对该家系的分子发病机制、临床预测进行了研究探讨,但具体机制尚待于进一步的体外表达研究。
